Reports: DNI651155-DNI6: Bridging the materials and pressure gaps in Rh-catalyzed syngas conversion
 printer friendly
printer friendly
 The
principal research objective of this project is to elucidate the role of high-surface
area oxide catalyst support in modulating the selectivity and activity rhodium
(Rh) catalysts in syngas (CO + H2) conversion to ethanol (EtOH). This is a variant of the conventional Fischer-Tropsch process, generating long-chain liquid hydrocarbon oxygenates
from gasified biomass / coal. Since thermodynamics favors the formation of
methane and water, generation of alternative (more useful) products (like
ethanol) must necessarily exploit subtle modifications of reaction kinetics enabled
by catalyst modification. Experimentally, it has been found that changes in the
oxide support from silica (SiO2) to titania
(TiO2) yields significant increases
in the production of C2+ oxygenates. Thus in this reaction, as in
many others, the support modulates activity either directly (by catalyzing
reactions on the oxide or at the nanoparticle/oxide interface) or indirectly
(by modifying the electronic structure of the metal nanoparticle itself, or
nanoparticle geometry.) The ultimate goal of our research is to understand the
detailed role of the oxide support in this specific
reaction, and also develop a more comprehensive understanding on support
effects in general.
The
principal research objective of this project is to elucidate the role of high-surface
area oxide catalyst support in modulating the selectivity and activity rhodium
(Rh) catalysts in syngas (CO + H2) conversion to ethanol (EtOH). This is a variant of the conventional Fischer-Tropsch process, generating long-chain liquid hydrocarbon oxygenates
from gasified biomass / coal. Since thermodynamics favors the formation of
methane and water, generation of alternative (more useful) products (like
ethanol) must necessarily exploit subtle modifications of reaction kinetics enabled
by catalyst modification. Experimentally, it has been found that changes in the
oxide support from silica (SiO2) to titania
(TiO2) yields significant increases
in the production of C2+ oxygenates. Thus in this reaction, as in
many others, the support modulates activity either directly (by catalyzing
reactions on the oxide or at the nanoparticle/oxide interface) or indirectly
(by modifying the electronic structure of the metal nanoparticle itself, or
nanoparticle geometry.) The ultimate goal of our research is to understand the
detailed role of the oxide support in this specific
reaction, and also develop a more comprehensive understanding on support
effects in general.
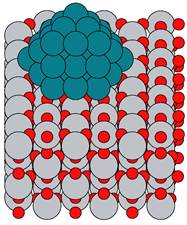
We have made significant progress towards these goals. In recent work (submitted to ACS catalysis) we developed realistic models of supported Rh nanoparticles, and used computational tools to examine the role of the metal-support interaction on reactivity. From experimental characterization data for Rh/SiO2 and Rh/TiO2 catalysts, we developed several representative catalysts model systems. We adopt a hemispherical three-layer cuboctahedral Rh37 nanoparticle model supported on silica and titania with differing surface terminations, characteristic of varying experimental conditions. Both supports are frequently utilized in experimental FT studies and are representative of an irreducible and a reducible oxide support, respectively. Based on energetic considerations, we focused on the rutile (110) face of titania and the (001) surface of α-quartz. We also examine hydroxylated variants of both surfaces, which may arise during operating conditions due to water dissociation (silica) or hydrogen spillover (titania). An example of the resulting “realistic” computational model is shown in Fig 1.
We found that addition of silica
support has little influence on adsorbate-metal binding energies when compared
to the unsupported nanoparticle, while a titania
yields considerable changes. Decoupling the electronic and geometric aspects of
the metal-support interaction, we find that geometric relaxation of the
nanoparticle due to the presence of the oxide support yields a fairly
systematic increase in adsorbate binding by ~0.1-0.3 eV,
independent of the nature of oxide, due to relaxation-induced strain and
increasing metal-metal bond distances. In contrast, the electronic contribution
to the metal-support interaction is far more sensitive to the composition and
termination of the oxide. We rationalized the latter in terms of charge
transfer between support and nanoparticle. Here, Lewis-neutral silica yields
little charge transfer, and thus little electronic support effect, whereas the
TiO2 and the hydroxylated TiO2
supports lead to a positive and a negative nanoparticle charging, respectively.
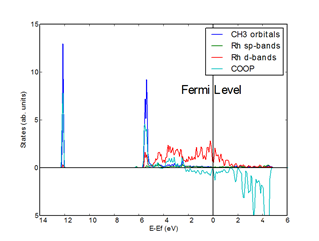
In turn, nanoparticle charging
 modulates
the adsorption energies by populating or depopulating states of anti-bonding adsorbate-metal
character around the Fermi level (see Figure 2).
modulates
the adsorption energies by populating or depopulating states of anti-bonding adsorbate-metal
character around the Fermi level (see Figure 2).
Crucially, these (sometimes significant) shifts in adsorbate binding energies induce corresponding changes to the reaction thermodynamics and kinetics for the elementary steps involving these adsorbed species; a straightforward thermodynamic analysis shows that changes in reaction enthalpy are directly related to the differential binding energy shifts of reactants and products. Based off of prior micro-kinetic modeling, we also showed that how the metal-support interaction can be exploited to influence the EtOH selectivity over methane: the overall selectivity toward EtOH production on the TiO2 support is due to the increased activation energy for the final hydrogenation step leading to methane, coupled with a small decrease in the activation energy for CH3 insertion into CO. In contrast, silica yielded only modest changes to these selectivity determining steps. These basic ideas provide a simple framework for considering the general influence of oxide supports in modulate catalyst activity and selectivity.
This work represents one of the few computational studies of a non-trivial reaction on a supported metal nanoparticle, and one of the first to comprehensively evaluate the role of the oxide support. The extensive calculations were facilitated from a grant of computer time on the NSF Teragrid. In addition to the above results, we have also collected preliminary data for an additional manuscript focusing on (i) the role of oxygen defects on TiO2 in modulating the supported nanoparticle; and (ii) the role of interfacial catalytic sites at the metal-support interface.
The bulk of this research has been conducted by Glen Jenness, a postdoctoral associate in my group who was funded via this grant. Although Glen arrived with a strong background in computational chemistry, he had little previous experience in catalysis. Over the course of this project, Glen acquired an expert mastery of the tools of computational heterogeneous catalysis. He intends to pursue this area more deeply, perhaps eventually at a national lab. To this end, he recently began a 2nd postdoc with Prof. Dion Vlachos (Delaware), a leader in this field.
This project has also been instrumental in kick-starting a new computational heterogeneous catalysis program within my own group. Based on the success of this project, we are pursuing additional (NSF) funding on a related project, focusing on the role of main-group promoters (e.g. Bi, Pb, Te) on enhancing the activity and selectivity of platinum-group metal alcohol oxidation catalysts. This project is based on a computational study of “realistic” catalysts model systems based on experimental characterization data, and thus shares many common themes with the above PRF-funded work.
Although not directly funded by this grant, Glen worked in close coordination with 4th year graduate student Benjamin Dunnington. Benjamin has developed a novel approach to developing “catalytic descriptors” that are based on the bonding of an adsorbate to a catalyst surface, and that allow us to analyze rigorous calculations in chemical intuitive terms. We hope to employ these catalyst descriptors in our future studies of support and promoter effects to provide enhanced “chemical” understanding of these complex phenomena.









