Reports: DNI150491-DNI1: [3,3]-Rearrangements of O-Vinyl Oximes: Stereoselective Synthesis of 1,4-Dicarbonyl Compounds
 printer friendly
printer friendlyThe specific aim of the proposed project was to develop the [3,3] rearrangement reactivity of O-vinyl oximes as a route to stereochemically defined 1,4-dicarbonyl compounds. The overall goal of the program is to explore and develop the chemistry of O-vinyl oximes as valuable precursors to privileged synthetic intermediates to streamline syntheses of pharmaceutically active molecules and desirable new materials. The specific aim of the project built on our preliminary studies that showed that a variety of O-propenyl oximes are easily accessible from O-allyl oximes via an iridium-catalyzed isomerization reaction and that O-propenyl oximes can be subsequently thermally converted to pyrroles. During the first and second year of the funding period, we discovered new rearrangements of O-alkenyl oximes and O-alkenyl hydroximates and developed a new copper-mediated coupling for the preparation oxime and hydroxylamine ethers, which provided a general, modular, and stereocontrolled route to our required starting materials. We also observed that an analogous copper-mediated coupling procedure provided direct access to N-alkenyl nitrones from fluorenone oxime and that these compounds undergo thermal rearrangements to form spiroisoxazolines. During the third year of the funding period we expanded our N-hydroxyphthalimide-mediated oxygenation chemistry to include the diastereoselective synthesis of α-oxygenated ketones and applied our copper-mediated etherification conditions to the preparation of O-vinyl benzophenone oximes which we used to access α-imino aldehydes through [1,3] rearrangements. In addition, we advanced the ketonitrone branch of our research program to include the synthesis of α,β-epoxyketimes through an copper-catalyzed oxygen-transfer rearrangement of N-aryl-α,β-unsaturated nitrones and the synthesis of dihydrocarbazoles through an addition and rearrangement reaction of N-aryl-α,β-unsaturated nitrones and allenes. Each of these four major advances in our research program are illustrated and described below.
I. Expansion of Boronic Acid Dioxygenation to Include the Diastereoselective Preparation of α-Oxygenated Ketones and the Preparation of Catechols from Aryl Boronic Acids
In year two of the research
program, we designed a method for the dioxygenation of alkenyl boronic acids
with N-hydroxyphthalimide using a two-step procedure involving an
initial C–O bond coupling between N-hydroxyphthalimide and an alkenyl
boronic acid followed by a subsequent [3,3] rearrangement which gave α-imidoyl
ketones that could be hydrolyzed to the corresponding α-hydroxy ketones.
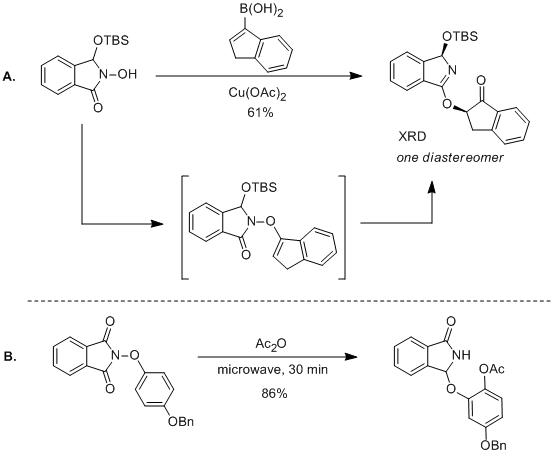
In year three, we discovered that N-hydroxy-3-siloxy-isoindolinones
could undergo a similar C–O bond coupling and an analogous diastereoselective
[3,3] rearrangement (Scheme 1A). The rearrangement of the isoindolinone
substrates is more facile than the phthalimide derivatives and the two-step
procedure occurs as a single-flask process at room temperature. This new diastereoselective
version of the dioxygenation process also tolerates tri-substituted alkenes and
allows for the preparation of tertiary carbinols. We are currently preparing a
manuscript discussing the scope of this transformation as well as preliminary
mechanistic studies. Further studies in this direction showed that N-aryloxy
phthalimides also undergo a [3,3] rearrangement to form substituted catechols.
We discovered that this transformation is most efficient when performed in a
microwave and can tolerate a same-flask acetyl protection (Scheme 1B). We
currently have more than 15 examples of this transformation and are exploring
the synthetic applications of this simple route to differentially protected
catechols.
Scheme 1.
II. Synthesis of α-Imino Aldehydes by the [1,3] Rearrangement of O-Vinyl Oximes
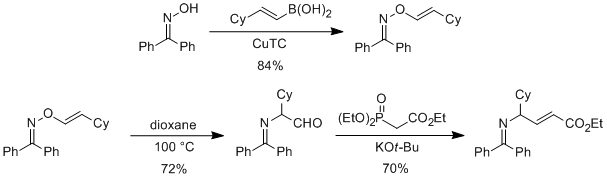
During year one of the research program, we observed that O-propenyl oximes undergo both [1,3] and [3,3] rearrangements to provide regiodefined pyrroles. By using the copper-mediated C–O bond coupling process discovered in year two, we were recently able to optimize a general route to O-alkenyl benzophenone oximes and showed that these compounds undergo thermal [1,3] rearrangements to provide α-imino aldehydes (Scheme 2). We determined that this thermal [1,3] rearrangement can be paired with a subsequent olefination process to give γ-imino-α,β-unsaturated esters in a single-flask procedure from O-alkenyl oximes. This work was recently published (Kontokosta, D.; Mueller, D. S.; Wang, H.-Y.; Anderson, L. L. Org. Lett. 2013, 15, 4830).
Scheme 2.
III. Preparation of N-Aryl α,β-Epoxyimines and Use in the Synthesis of Highly Substituted and Stereodefined Heterocycles
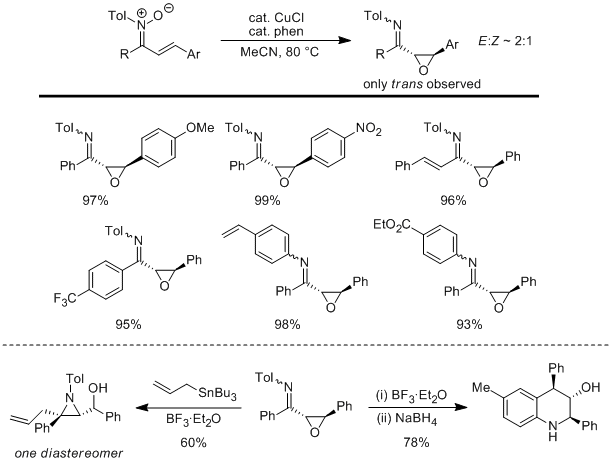
In year two of the research program, we discovered that copper-mediated C–N bond coupling can be used to form N-alkenyl nitrones from fluorenone oxime and alkenyl boronic acids. During year three, we used a similar approach to form N-aryl α,β-unsaturated ketonitrones from chalcone and dibenzylidene acetone-derived oximes. This method is important due to the fact that these ketonitrones are inaccessible by known methods and development of this methodology allowed us to explore the reactivity of these interesting compounds. As shown in Scheme 3, we discovered these α,β-unsaturated N-aryl nitrones undergo a copper-catalyzed oxygen transfer rearrangement to give α,β-epoxyketimines which are inaccessible by condensation and undergo chemoselective reductions as well as stereoselective addition and rearrangement reactions to give highly substituted and stereodefined tetrahydroquinolines and aziridines (Mo, D.-L.; Anderson, L. L. Angew. Chem. Int. Ed. 2013, 52, 6722). As shown in Scheme 4, we also discovered that N-aryl nitrones undergo addition and rearrangement reactions with electron-
Scheme 3.
Scheme 4.
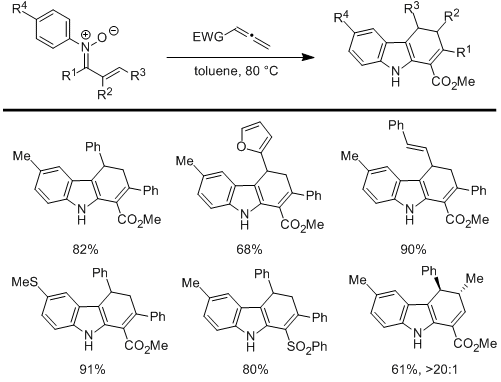
deficient allenes to give dihydrocarbazoles. This exciting cascade process provides a new, efficient, and diastereoselective approach to tetrahydrocarbazoles which have a history of being both important and challenging synthetic targets. We have recently submitted a manuscript describing the scope and generality of this new method (Mo, D.-L.; Bhardwaj, C.; Wink. D. J.; Anderson, L. L. submitted).
Summary.
In summary, funding for this project has allowed us to not only explore the reactivity of O-vinyl oximes, but also the reactivity of related O-alkenyl hydroximates, N-alkenyl ketonitrones, and N-aryl ketonitrones, and allowed us to demonstrate how these uncommon compounds can be easily prepared from simple starting materials and used as valuable synthetic intermediates for the preparation of challenging C–O, C–N, and C–C bonds. The diversity of the N–O bond rearrangements that we have discovered and applied to important synthetic problems over the past three years has laid a foundation for our research program and will continue to be pursued with other funding sources.









