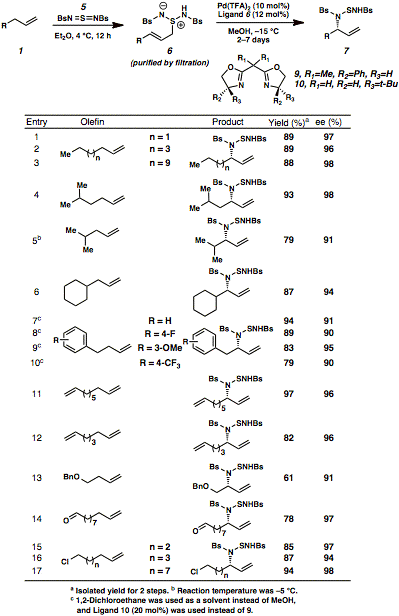Reports: DNI151710-DNI1: Enantioselective Allylic Amination of Olefins
 printer friendly
printer friendlyChemists havedeveloped many powerful strategies of synthesizing chiral molecules, which hasimproved our quality of life in the form of pharmaceutical drugs and syntheticmaterials. Unfortunately, the generation of these useful chemicals takes aheavy economic and environmental toll. Therefore, the development of catalyticenantioselective strategies that can reduce the monetary and ecological costsof chemical synthesis is an active area of research. In our ACS-PRF grantapplication, we proposed the enantioselective synthesis of medicinally valuablechiral amines via metal-catalyzed allylic amination of simple olefins, whichare common components of petrochemical feedstock and are attractive startingmaterials for the generation of complex molecules. We have made significantprogress towards achieving this goal.
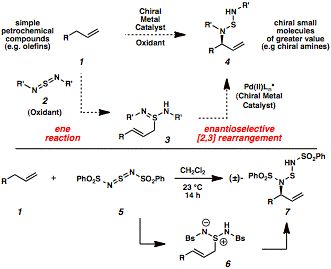
Our strategyfor an economically and environmentally friendly generation of chiral amines isbased on a two-step oxidative process (Scheme 1). In the first step an olefin reacts with sulfur-based oxidant2 to generate ene adduct 3. Thisintermediate is then converted in the second step into chiral amine 4 through a metal-catalyzed enantioselective [2,3]rearrangement.
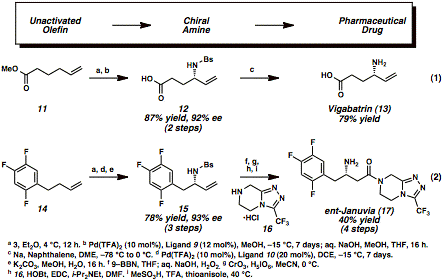
Our approachwas inspired by the work of Sharpless, Kresze, and Katz, who reported over 30years ago that an olefin 1 reacts witharylsulfonyl sulfurdiimide reagent 5 via a hetero-ene reaction to generate zwitterionic species 6 (Scheme 1). This reactive intermediate undergoes aspontaneous [2,3]-rearrangement, which results in the formal allylic aminationof the olefin starting material (1to 7). Our key insight into thischemistry was that this rearrangement could be controlled by a chiralpalladium(II) catalyst, which accelerates the enantioselective[2,3]-rearrangement of zwitterion 6to allylic amine derivative 7. Our initialexperiments focused on the identification of a high oxidation state nitrogensource that would couple with a terminal olefin to form an ene adduct, such as 1, which would not undergo a background [2,3]-rearrangementin the absence of a chiral catalyst. This proved challenging, since mostimido-sulfur and imido-selenium oxidants that are suitable for allylicamination strategies participate in uncatalyzed thermal ene reactions followedby [2,3]-rearrangements. For example, the early work of Sharpless and Kreszehighlighted the racemic allylic amination of terminal olefins at roomtemperature with arylsulfonyl sulfurdiimide reagents through the two-steppericyclic reaction sequence. When wereexamined these arylsulfonyl sulfurdiimide oxidants, we discovered thatbenzenesulfonyl sulfurdiimide 5(BsN=S=NBs is C6H5SO2N=S=NSO2C6H5)and 1-octene 1a formed stableene adduct 6a at 4 °C in highyield (Scheme 2). This zwitterionic intermediate was purified by simplefiltration without any chromatography and did not rearrange at temperaturesbelow 0 °C for multiple days (entry 1). Coupling between nitrogen-containingoxidant 5 and terminal olefinsat low temperatures was therefore selected as the platform for our catalyticenantioselective allylic amination of olefins. In our earlyattempts to accelerate the [2,3]-rearrangement of zwitterion 6a, palladium(II) salts such as Pd(OAc)2were promising catalysts for the desired rearrangement (entry 2). After anextensive screening of reaction parameters (including solvent, chiral ligand,and palladium source), we identified Pd(TFA)2 and bisoxazolineligand 9 as the optimal catalystcomplex for generating allylic sulfonamide product 7a in 89% yield and 96% enantiomeric excess (ee) atÐ15 °C in anhydrous methanol (entry 3). The overall two-step protocol onlyrequires one chromatographic purification, which simplifies the operation ofthis reaction on large scale. Havingidentified optimized reaction conditions for the enantioselective conversion ofolefins into allylic amines, we explored the substrate scope of this process ona preparative scale (2-4 mmol). Table 1 highlights the compatibility of thereaction with a wide range of functional groups, including non-directing simplehydrocarbon chains (entries 1-3), hindered branched hydrocarbons (entries 4-6),aromatic rings with different substitution patterns (entries 7-10),polyunsaturated hydrocarbons (entries 11-12), ethers (entry 13), aldehydes(entry 14) and primary alkyl chlorides (entries 15-17). Thisenantioselective reaction has potential for converting olefins intobiologically active nitrogen-containing chiral molecules, which is exemplifiedby the synthesis pharmaceutical drugs (Scheme 3). Commercially availableunsaturated ester 11 was converted intoenantioenriched antiepileptic drug Vigabatrin (Equation 1). While a racemic version of the allylicamination process has been reported for the synthesis of this drug, we wereinterested in developing an asymmetric synthesis since only one enantiomer ofVigabatrin is pharmacologically active. Our enantioselective allylic aminationprotocol yielded chiral sulfonamide 12, which was then deprotected to reveal the enantioenriched HCl salt ofVigabatrin (13). Trifluorophenylbutene 14 was similarly converted intoJanuvia (Equation 2), an FDA-approved DPP-4 inhibitor for the treatment of TypeII diabetes. Olefin 14 wasinitially treated with benzenesulfonyl sulfurdiimide 5 (BsN=S=NBs) and the chiral palladium catalystderived from Pd(TFA)2 and bisoxazoline ligand 10 to produce chiral sulfonamide 15 after treatment with aqueous base. Thisintermediate was then transformed into the free base of ent-Januvia(17) through a short sequence of steps. The synthesesof Vigabatrin and Januvia highlight the potential utility of enantioselectiveallylic amination as an economically efficient and environmentally benignalternative for the production of pharmaceutical drugs. In addition, thismethodology may spur additional drug development because of the accessibilityof terminal olefins. We arecurrently interested in gaining more mechanistic insight into the mode ofstereoinduction in the enantioselective rearrangement step while alsoincreasing the functional group compatibility of the transformation. Weanticipate that this method will be widely useful for the synthesis ofnitrogen-containing pharmaceutical agents from inexpensive and abundantunactivated olefins. Moreover, our ability to control the stereoselectiverearrangement of reactive zwitterions with chiral catalysts may have broaderimplications for the conversion of other simple starting materials into chiraland medicinally valuable products. Our discoveryof an enantioselective allylic amination of olefins represents a conceptualframework for the generalized stereoselective functionalization of inert CÐHbonds via enantioselective rearrangements. In the immediate future, we willtackle the more transformative problem of converting unactivated CÐH bonds inunsaturated hydrocarbons into any functionalized bond (CÐC, CÐO, and CÐhalogen)via asymmetric rearrangements, with a broader goal of changing the ways inwhich pharmaceutical drugs and other functional materials are produced. Table 1 <>