Reports: DNI652101-DNI6: Understand Methane Hydrates Formation through Accelerated Molecular Simulations
 printer friendly
printer friendlyThe objective of the project is to overcome the current limits in the theoretical understanding of methane hydrates, i.e., insufficient sampling and large gap in conditions between simulations and experiments, through employing accelerated molecular simulation approaches. To solve these problems, we proposed to combine the high-efficiency sampling approach—forward flux sampling (FFS) method1, with the coarse-grained water model2. Along this research line, significant progresses have been achieved during the first year of the project, as summarized below.
The foremost step in applying FFS approach is to develop a robust order parameter that allows characterizing and driving nucleation. Recognizing that hydrates are distinguished from ice and water in their unique structural signature of polyhedral cages 5126n (n=0, 2, 3, and 4), we have developed a strategy that focuses particularly on the formation of various polyhedral cages, and less on the stacking order of the cages.
The order parameter λ we have developed has the following features that are crucial for the understanding of methane hydrate nucleation: (1) it does not bias towards the formation of any particular hydrate phase. Indeed the formation of sI, sII and amorphous phases is all implied; (2) it allows various nucleation pathways to compete, which is the key for establishing and validating nucleation mechanisms; (3) the computational cost is reasonable. The order parameter was successfully implemented in our MD code.
It is a primary goal of the project to obtain the rate of hydrate nucleation via molecular simulations. This goal has been achieved through combining FFS approach, mW water model, and the developed order parameter. In FFS3, the rate is expressed as RAB=Φλ0 P(λn|λ0) where Φλ0 is the flux rate reaching the first interface λ0 from basin A and P(λn|λ0) is the probability for a trajectory that starts from λ0 and eventually reaches B. The computation of the growth probability plays the pivotal role in acquiring the total rate, and its convergence is shown in Fig. 1. To best of our knowledge, this is the first time that the nucleation rate of hydrates has been computed explicitly through molecular simulations.
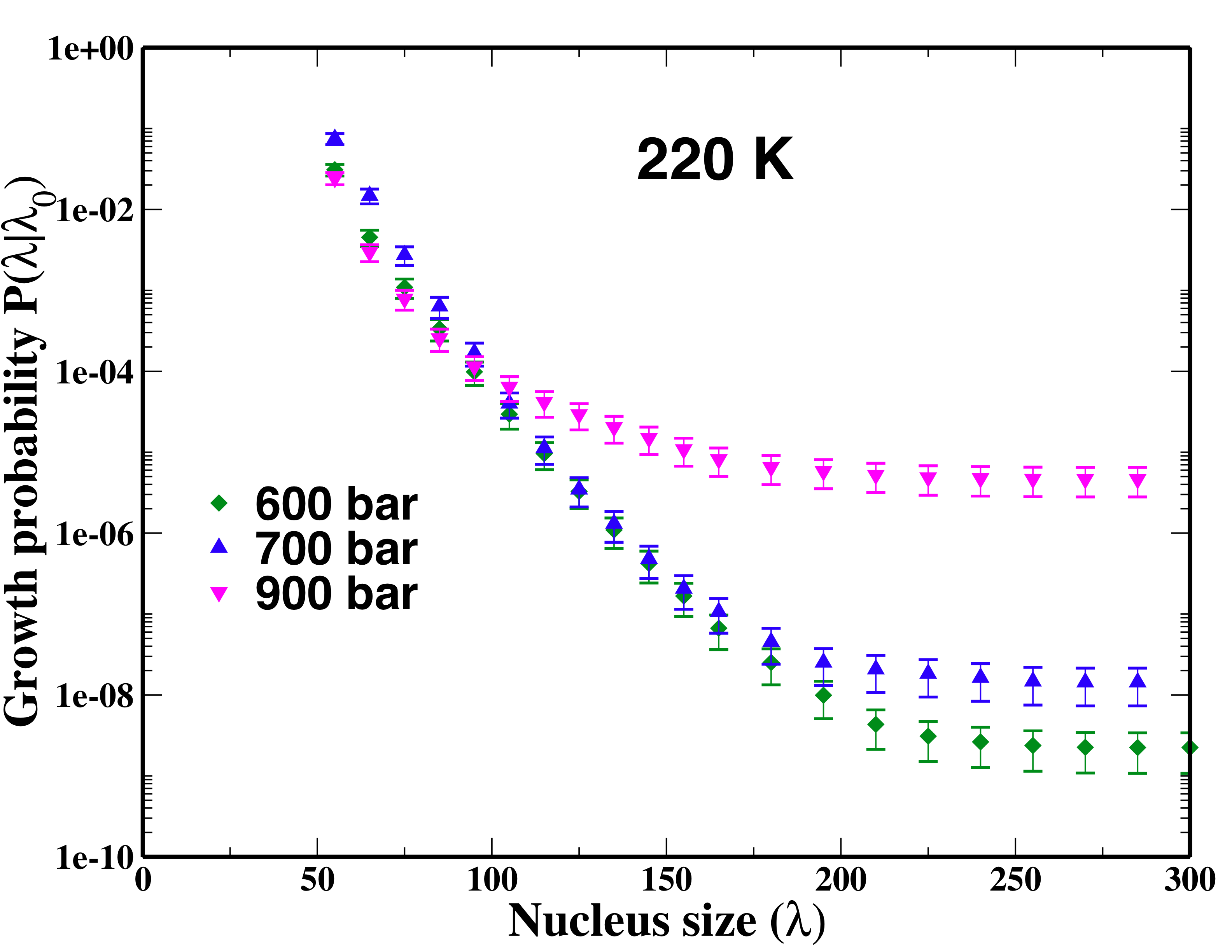
Two fundamental questions are raised concerning the kinetic relevance of the formation of ice and hydrates: Which crystal would form first at a given condition? Would the crystal initially formed transform into the other? The first question can now be addressed by comparing explicitly the computed nucleation rates of ice and hydrates under the same condition. As the formation of hydrate typically involves high pressure, our research along this line has been initially focused on the pressure effect of nucleation rates in both crystals. We began our investigation by exploring ice nucleation under pressure, which finds its relevance in the experiments aiming to probe water's behavior in its “no man's land” by supercooling water nano droplets. The internal pressure within water nano droplets, i.e., Laplace pressure, is of the magnitude (a few hundred bars) comparable to that under which hydrate forms.
For the case of ice nucleation within nano droplets, we found its nucleation rate decreases with the radius of the droplets, which was attributed to the increased Laplace pressure within the droplets when size is decreased4. The computed ice nucleation rate decreases monotonically with the pressure within the range of study, which is related to the change of nucleation barrier due to the contribution of pΔv term (where p is the pressure, Δv is the molar volume difference between water and ice).
For the case of hydrate, we find pressure plays a more complex role on the nucleation rate. While the exact cause is still under investigation, we expect it to be related to the more complex factors controlling the formation and aggregation of cages, which adversely depend on pressure. Regardless such dependence, we find at 220 K, the nucleation rate of methane hydrate is always higher than that of ice. The next question to address is whether cross nucleation between ice and hydrate is kinetically possible.
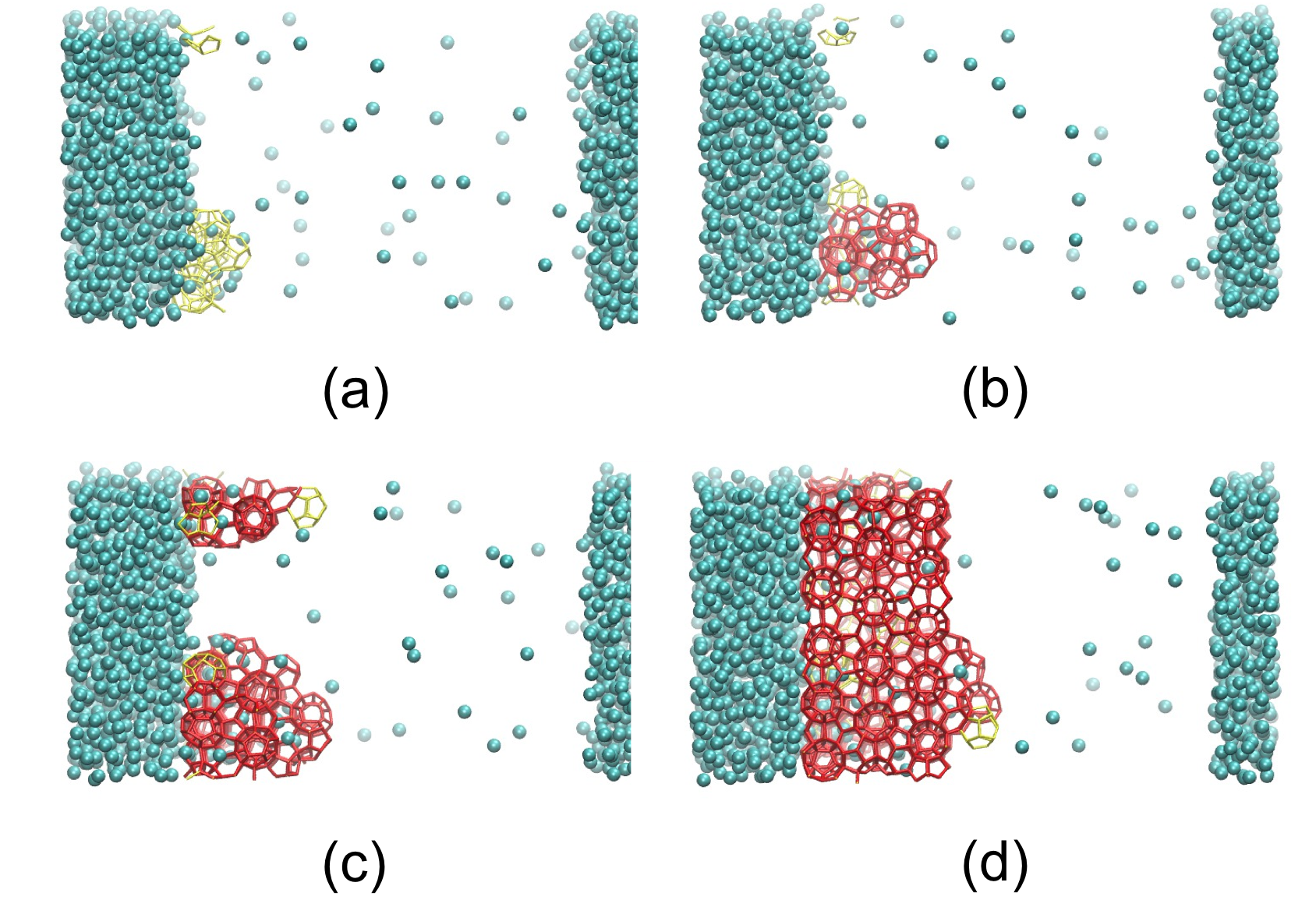
The large ensemble of nucleation trajectories acquired from FFS sampling also allows identifying multiple hydrate nucleation pathways. We find a hydrate nucleus may possess amorphous core, as formerly discovered by previous studies, but may also display crystalline ordering, which has not been reported. Regardless the initial ordering the hydrate cages, both types of hydrate nuclei will yield crystalline hydrates during the subsequent growth process. Fig. 2 shows an initially amorphous hydrate cluster becomes crystalline hydrate during the growth process, at 215K and 100 bar. It is our next step to understand what factors control the nucleation pathways, and whether it would be possible to control the polymorphism of hydrate crystals.
 |
The ACS PRF DNI award has provided valuable support allowing the PI to open a new research direction. The project has been supporting a postdoctoral scientist and involving a graduate student.
References:
ADDIN PAPERS2_CITATIONS <papers2_bibliography/>1. Allen, R. J., Frenkel, D. & Wolde, Ten, P. R. Simulating rare events in equilibrium or nonequilibrium stochastic systems. J Chem Phys 124, 024102 (2006).
2. Jacobson, L. C. & Molinero, V. A Methane-Water Model for Coarse-Grained Simulations of Solutions and Clathrate Hydrates. J Phys Chem B 114, 7302–7311 (2010).
3. Allen, R. J., Frenkel, D. & Wolde, Ten, P. R. Forward flux sampling-type schemes for simulating rare events: Efficiency analysis. J Chem Phys 124, 194111 (2006).
4. Li, T., Donadio, D. & Galli, G. Ice nucleation at the nanoscale probes no man's land of water. Nat Commun 4, 1887 (2013).









