Reports: DNI552279-DNI5: Rapid Prediction of Sulfide and Oxide Catalyst Surface Structures Using Insight from Density Functional Theory
 printer friendly
printer friendlyIntroduction. Metal-sulfides, specifically Ni and Co promoted MoS2, are the industrial hydrotreating catalysts of choice and are particularly active for hydrodesulfurization (HDS). Recently, it was shown that sulfur vacancy formation energies can serve as reactivity descriptors for HDS catalysis, when they are calculated for realistic surface coverages under hydrotreating reaction conditions.1 Realistic surface coverages can be calculated using an ab-initio thermodynamics approach resulting in a surface phase diagram, but this approach is time consuming and not suited for computational catalyst screening.2 Similarly, metal-oxides are good candidate materials for the related hydrodeoxygenation (HDO) process, which is significantly less studied, but conceptually similar to HDS. The overarching goal of this project is to gain fundamental insight from density functional theory (DFT) calculations to develop approximate models that allow for a rapid prediction of the active catalyst surface of sulfides and oxides under reaction conditions and enable rapid computational screening for hydrotreating processes.
Preliminary Progress. While MoS2-based catalysts are industrially used for economic reasons, RuS2 has in fact the highest HDS activity.1 Therefore, we initially investigated the RuS2(111) surface to derive a thermodynamic phase diagram from DFT simulations, which is shown in Figure 1. However, the surface structure of the RuS2(111) surface is rather complex and can exhibit five different surface terminations.3 While this can be treated within the ab-initio framework to predict the large number of preferred surface terminations and coverages under HDS conditions, this surface is not well suited as starting point for the derivation of a rapid prediction method based on thermodynamic surface descriptors.
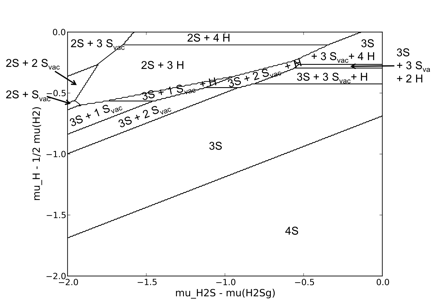
Figure 1 RuS2(111) surface phase diagram. The nomenclature for possible surface terminations is according to reference [3].
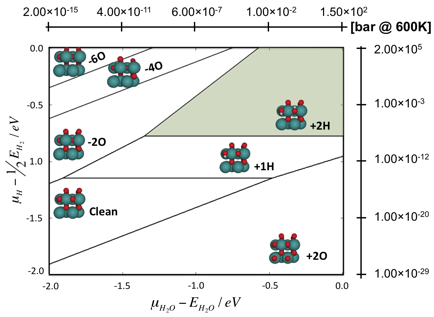
Figure 2 Surface phase diagram for RuO2(110).
The oxygen analogue of RuS2 is RuO2, which preferentially exposes its rutile RuO2(110) surface. This surface is comparatively simple and has only two types of surface oxygen atoms, i.e. bridging and 3-fold coordinated oxygen sites. The surface phase diagram of RuO2(110) as predicted from DFT is shown in Figure 2, and in the interesting range of hydrotreating processes conditions, the surface is partially reduced with hydroxyl groups in the bridging positions. Based on the DFT calculated surface free energies of the RuO2(110) surface, we proposed a formula to approximate the surface free energy using only the binding energies of hydrogen and the oxygen vacancy formation energies at both surface oxygen sites (bridging, 3-fold) as input parameters. The model was semi-successful, but needed to be refined with interaction parameters between nearest neighbor vacancy sites and/or hydroxyl groups. The improved model uses an additional ten adjustable interaction parameters and can accurately reproduce the DFT calculated surface free energy over the complete range of vacancy and hydroxyl coverages as shown in Figure 3.
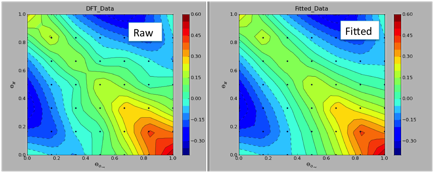
Figure 3 Oxygen vacancy and hydrogen coverage dependent surface free energy from raw DFT data (left) and using a parameterized model (right).
Consequently, the RuO2(110) phase diagram derived from the simplified model is in very good agreement with the phase diagram using the full DFT results. Based on this success for RuO2, we have repeated this approach for 8 different rutile(110) surfaces, and achieved similarly good results. Next, we tested if our model can also be applied for doped surfaces and studied Ru-doped TiO2. Two doped structures were investigated: (i) a single Ru atom in the TiO2(110) surface, and (ii) a monolayer of RuO2(110) on TiO2(110). In both cases the simplified description of the surface free energy resulted in nearly identical phase diagrams as the full ab-initio approach.
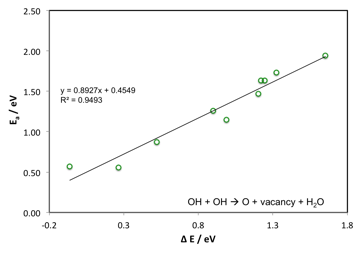
Figure 4 Scaling relationship for OH disproportionation.
Furthermore, we considered the vacancy formation kinetics on rutile (110) surfaces by investigating the activation barrier for the disproportionation reaction of two neighboring surface hydroxyl groups leading to the desorption of H2O. The results are given in Figure 4 and show a strong correlation with the enthalpy change of vacancy formation. Other oxygen vacancy formation pathways are also being considered in our ongoing work.
Summary and Future Work. Very promising results for the rapid prediction of rutile metal-oxide surfaces under realistic reaction conditions have been obtained, and we anticipate that the methodology, once fully developed, can be applied to a much wider range of metal-sulfide and metal-oxide surfaces. The next class of materials we study will be MoS2-type metal-sulfides and we plan to search for common thermodynamic descriptors that allow to translate results from oxides to sulfides and vice versa. Especially interesting for catalytic screening studies are the facts that vacancy formation kinetics are correlated with their formation energy and that the approach is also applicable to doped materials. The above results will be published in the near future with my ACS-PRF supported graduate student Byeongjin Baek as the first author. He passed his second year qualifying exam with flying colors and has presented his results at the CAMd Summer School at DTU in Denmark and at the Gordon Research Seminar and Conference in Switzerland. I have included his results in a total of four invited and contributed talks, including a presentation in the final round for the Gerhard Ertl Young Investigator Award, and won the best poster award at the 528. Wilhelm and Else Heraeus Seminar in Germany. The results were also included in several related proposals and most recently, in the DOE Early Career Award. I expect our work to have a large fundamental impact on the computational design of improved catalytic materials for hydrotreating processes and this ACS-PRF award has been instrumental for the launch of this project.
References
1. P. G. Moses, L. C. Grabow, E. M. Fernandez, B. Hinnemann, H. Topsze, K. G. Knudsen and J. K. Nzrskov, "Trends in hydrodesulfurization catalysis based on realistic surface models", Journal of Catalysis (2013) submitted.
2. L. C. Grabow, "Computational Catalyst Screening", in Computational Catalysis. Eds.: A. Asthagiri and M. Janik. RSC Publishing 2013.
3. D. R. Alfonso, "Computational Investigation of FeS2 Surfaces and Prediction of Effects of Sulfur Environment on Stabilities", The Journal of Physical Chemistry C 114 (2010) 8971–8980.









