Reports: DNI152252-DNI1: Remodeling C-H Bonds: Selective Catalysis of Hydrogen Atom Transfer
 printer friendly
printer friendlyHydrogen atom transfer catalysis mediated by ammonium phenoxyls Our original ACS PRF proposal outlined the use of persistent phenoxyl radicals as catalysts for enantioselective hydrogen atom transfer processes. These oxidants are known in many contexts to homolytically activate C-H bonds, and also to serve as hydrogen
| Figure 1. Phenoxyl-mediated HAT catalysis
|
 atom donors for the reductive quenching of
carbon-centered radicals. We proposed that by merging these processes, it may
be possible to develop new radical redox chemistries wherein radical
intermediates are first generated via HAT to the phenoxyl acceptor, then react
further to form a new radical intermediate that can re-abstract hydrogen from
the phenol form of the catalyst to close the catalytic cycle (Figure 1).
atom donors for the reductive quenching of
carbon-centered radicals. We proposed that by merging these processes, it may
be possible to develop new radical redox chemistries wherein radical
intermediates are first generated via HAT to the phenoxyl acceptor, then react
further to form a new radical intermediate that can re-abstract hydrogen from
the phenol form of the catalyst to close the catalytic cycle (Figure 1). Extensive efforts to develop these ideas were met with mixed results. C-H abstraction events were realized for some substrates classes, and significant primary KIE values were often observed, consistent with a rate-determining HAT activation. However, due to polar effects, these highly electrophilic ammonium phenoxyls tended to react most readily with C-H bond possessing some hydridic character, such as a-oxy or a-amino C-H bonds. Upon abstraction, such radicals are typically quite reducing, and we observed that they would typically engage in rapid electron transfer reactions with an additional equivalent of the persistent phenoxyl to generate cationic intermediates and closed shell phenols which were no longer capable of participating in the desired reaction manifolds. Extensive experimentation and supporting mechanistic studies ultimately were not able to overcome this limitation, prompting us to evaluate new approaches to the proposed research.
Proton-coupled electron transfer In seeking alternative approaches to the proposed research, we became interested in the synthetic viability of concerted proton-coupled electron transfer (PCET) reactions to serve
| Figure 2. Energetic advantages of PCET activation
|
 surrogates for as traditional
hydrogen atom transfer processes. PCET mechanisms are unconventional redox
processes in which chemically independent electrons and protons are
simultaneously exchanged in a single elementary step, in contrast to HAT
processes where the proton and electron travel together. The energetic coupling enforced by a
shared transition state allows a favorable driving force associated with one
exchange event to compensate for unfavorable energetics in the other without
requiring the generation of an intermediate. The benefits of this thermodynamic
compensation are often manifested kinetically, allowing concerted PCET to
proceed at rates significantly faster than either competing stepwise transfer
pathway (Figure 2A). In turn, this enables facile radical generation using
redox catalysts or reagents with potentials far less energetic (often >1 V)
than those of their substrates.
surrogates for as traditional
hydrogen atom transfer processes. PCET mechanisms are unconventional redox
processes in which chemically independent electrons and protons are
simultaneously exchanged in a single elementary step, in contrast to HAT
processes where the proton and electron travel together. The energetic coupling enforced by a
shared transition state allows a favorable driving force associated with one
exchange event to compensate for unfavorable energetics in the other without
requiring the generation of an intermediate. The benefits of this thermodynamic
compensation are often manifested kinetically, allowing concerted PCET to
proceed at rates significantly faster than either competing stepwise transfer
pathway (Figure 2A). In turn, this enables facile radical generation using
redox catalysts or reagents with potentials far less energetic (often >1 V)
than those of their substrates. These energetic characteristics have important implications for synthesis. As in HAT, the thermodynamics of a PCET reaction can be described by the difference in bond dissociation free energies (BDFE) between two bonds undergoing exchange. While no bond in the acid-reductant pair formally undergoes homolytic cleavage, a free energy fully equivalent to a BDFE may be calculated for any acid/reductant pair using a thermodynamic cycle comprised of the pKa's and redox potentials of its constituents, as well as a constant that accounts for both proton reduction and solvation terms (Figure 2B). This formalism enables the thermodynamics of any proposed PCET event to be evaluated by comparing the formal BDFE of a given acid/reductant pair to the strength of the new bond formed in the transfer event, or vice versa for oxidative processes.
Excitingly, PCET mechanisms present thermodynamically viable pathways for catalytic radical generation from nearly any common organic functional group. We have identified compatible base/oxidant combinations competent to abstract hydrogen from bonds with bond-dissociation free energies (BDFEs) >100 kcal/mol. (Figure 2C) Additionally, we have established acid/reductant combinations that can serve as formal hydrogen atom donors capable of forming bonds weaker than 20
| Figure 3: PCET enabled catalytic ketyl olefin coupling
|
 kcal/mol, (which is 30 kcal/mol weaker
than what is currently thermodynamically possible using known unimolecular
H-atom donor catalysts). This enormous dynamic range suggests that we will be
able to dramatically expand the scope of known radical generation technologies
(and formal HAT catalysis).
kcal/mol, (which is 30 kcal/mol weaker
than what is currently thermodynamically possible using known unimolecular
H-atom donor catalysts). This enormous dynamic range suggests that we will be
able to dramatically expand the scope of known radical generation technologies
(and formal HAT catalysis).We have recently demonstrated the viability of these ideas in the development of a new catalytic protocol for ketyl olefin coupling (Figure 3). In these reactions, kinetic studies demonstrated the lowest energy pathway to ketyl radical formation required electron transfer from a Ru- or Ir-based redox catalyst to a ketone acceptor to occur in concert with proton transfer from a phosphoric acid through the agency of a hydrogen bond. This PCET process forms a new neutral ketyl intermediate with a remarkably weak O-H bond (26 kcal/mol) that would be energetically inaccessible using any known unimolecular hydrogen atom donor. These intermediates were found to cyclize readily onto pendant olefins, forming useful annulated products under unprecedentedly mild catalytic conditions.
Additionally, concerted PCET activation presents unique opportunities for the development of enantioselective radical transformations. By kinetically coupling the electron transfer event to substrate binding by the Brznsted acid or base, PCET ensures that radical intermediates are exclusively generated as catalyst-bound
| Figure 4: Asymmetric PCET enabled azapinacol couplings
|
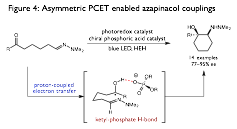 adducts. For ketyls, we have found that these
successor hydrogen bonds formed after PCET between the conjugate base of a
chiral phosphoric acid and the neutral radical are remarkably strong and can
persist throughout the course of subsequent bond forming events, establishing a
basis for asymmetric induction (Figure 4). We have successfully exploited this
observation to develop the first catalytic enantioselective aza-pinacol
coupling between ketones and hydrazones, furnishing valuable vicinal amino
alcohol products from simple starting material with excellent levels of
diastereo- and enantioselectivity (Figure 4). We anticipate these findings will
be applicable to many additional substrate classes as well, and that concerted
PCET will prove to be a general synthetic strategy for carrying out
enantioselective radical chemistries in a catalytic manifold. A manuscript describing this work is in preparation.
adducts. For ketyls, we have found that these
successor hydrogen bonds formed after PCET between the conjugate base of a
chiral phosphoric acid and the neutral radical are remarkably strong and can
persist throughout the course of subsequent bond forming events, establishing a
basis for asymmetric induction (Figure 4). We have successfully exploited this
observation to develop the first catalytic enantioselective aza-pinacol
coupling between ketones and hydrazones, furnishing valuable vicinal amino
alcohol products from simple starting material with excellent levels of
diastereo- and enantioselectivity (Figure 4). We anticipate these findings will
be applicable to many additional substrate classes as well, and that concerted
PCET will prove to be a general synthetic strategy for carrying out
enantioselective radical chemistries in a catalytic manifold. A manuscript describing this work is in preparation.Lastly, we are working to develop oxidative PCET processes in which oxidant/base pairs homolytically activate strong C-H and N-H bonds, offering new approaches to catalytic radical generation and further opportunities for synthesis. We appreciate the PRF's support of these research endeavors, which have had a significant positive impact on our group









