Reports: ND150715-ND1: Organocatalytic Tether Formation
 printer friendly
printer friendlyIntroduction
Catalysis of intermolecular reactions is at the basis of chemical reactivity. Many of the most effective catalysts perform substrate activation while favoring substrate preassociation. Such pre-organization of reagents typically results in increased activity and selectivity, since in this fashion the catalyst helps minimize the entropic penalty associated with intermolecular reactions. Not surprisingly, multiple transformations building on preassociation have been developed, and several families of bifunctional catalysts have emerged. However these catalysts typically use weak interactions to perform preassociation (e.g. H-bonding). In contrast, the catalysis of transformations only through temporary intramolecularity has received little attention. This is surprising since rate accelerations of 104-108 for 1 M reactants can be obtained via temporary intramolecularity. Indeed, simple catalysts operating only through this pathway are rare, perform relatively simple reactions and highly efficient asymmetric catalysts had not been reported.
In 2011, our group reported the use of simple aldehydes as tethering catalysts. Building on Knight's seminal work, we showed that selected aldehydes catalyze intermolecular Cope-type hydroaminations via temporary intramolecularity. As illustrated below, the reaction proceeds via in situ formation of a mixed aminal (I), which allows for a facile intramolecular hydroamination event. As opposed to traditional tethering strategies, this catalytic method does not require additional synthetic steps for the installation and cleavage of the tether. Furthermore, enantioenriched molecules can be formed using chiral aldehydes (up to 97%ee; reported in 2013). Following our communication we have gained solid mechanistic understanding, which includes the rate law of the reaction and identification of two catalyst inhibition pathways (reported in 2012). This work also showed why catalysts possessing a destabilized ground state are more reactive: they favor the required preassociation event, and this Keq is in the rate law of the reaction. This understanding led to the identification of an improved catalyst, formaldehyde, which is more active and allows more difficult hydroaminations to proceed. In addition, we have identified a bicyclic chiral catalyst (94%ee), which is remarkably robust toward epimerization.

Progress report
The purpose of this Grant was to expand this catalytic tethering approach to other reactions than hydroamination. The goals of the original proposal fell into three categories: catalyst optimization, reaction discovery and enantioselective catalysis. The first objective was to provide an in-situ, catalytic alternative to known tethered strategies. The second objective was the development of novel reactivity, including reactions relying on concurrent tandem catalysis. The third and final objective was to explore stereoselective variants using a chiral organocatalyst. As described below, solid progress has been made.
Efforts directed toward the development of new reactivity were discussed in the progress report submitted in 2012. These efforts led to the reinvestigation a hydrolysis system pioneered by Commeyras to gain solid footing toward new reactivity. Over 25 years ago, Commeyras developed various carbonyl directed hydrolysis reactions via hemiaminal intermediates. Hydrolyses of α-amino-esters, -amides, -thioamides and -nitriles were reported. However, these reactions used stoichiometric amounts of a carbonyl catalyst (even though turnover should occur), and the highest enantioselectivity reported is 42%ee. An illustrative example is the formaldehyde "catalyzed" hydrolysis of α-aminoamides (Eq 1).
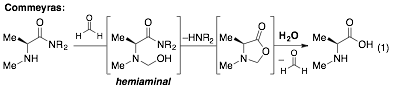
This work is important from a tethering standpoint even if the "tethered nucleophile" can only be water and if the issue of catalytic activity (turnover frequency and rate) has not been addressed. Our efforts in this system have proved rewarding (Eq 2): 1) a efficient version using catalytic amounts of the carbonyl compound and of the base has been developed (also constituting results on the concurrent tandem catalysis objective); 2) reactivity with various carbonyl catalysts mirrors what is observed for hydroamination, indicating that destabilized aldehyde catalysts are optimal to promote preassociation (favorable pre-equilibrium); 3) formaldehyde is an optimal catalyst for the transformation (i.e. a more efficient "tethering" system has been identified).

Synthetically, this allows the hydrolysis of "Strecker adducts" under mild conditions via the use of temporary intramolecularity. The efficiency of this procedure prevents the cyanohydrin decomposition that typically occurs under basis conditions, and provides an alternative to the forcing acidic conditions that are routinely used to hydrolyze Strecker adducts. The results shown above were well received at the 2013 Gordon Conference on Organic Reactions and Processes, and a manuscript will be submitted for publication shortly. This work will also provide the understanding that is currently lacking in the field of carbonyl catalyzed hydrolysis reactions.
Origin of life debate. This work will also contribute to the origin of life debate. The Miller-Urey "primordial soup" experiments showed that a-amino acids were produced under conditions mimicking the primitive atmosphere present on Earth. These conditions rely on the hydrolysis of Strecker adducts, and our work highlights that a key building block for organic molecules, formaldehyde, is also an efficient catalyst for hydrolysis reactions. Activity was also observed for C2-C4 aldehydes. Overall, this work addresses two important issues: 1) it shows the catalytic efficiency of basic aldehydes to perform hydrolyses reactions; 2) it provides an activation manifold addressing the "low concentration problem" inherently linked to chemical evolution via intermolecular reactions.
Asymmetric catalysis. Our hydroamination studies have shown that are that highly effective asymmetric catalysis is possible, and that the stereochemical integrity of the bicyclic aldehyde catalyst is excellent under basic conditions. The need to achieve kinetic control during the preassociation event remains a challenge for asymmetric hydrolyses reactions, since thermodynamic control should result in a poor selectivity during the formation of a chiral tether and, consequently, to poor enantioselectivity in the products. While efforts to achieve a highly enantioselecive variant of this reactivity have not been fruitful to date, the understanding gained is very useful and work is ongoing...
Future Plans and Outlook
In summary, this New Directions grant will shortly yield a communication on aldehydes as efficient catalyst for hydrolysis reactions. This work will highlight the synthetic potential of catalytic approaches using the temporary intramolecularity associated with in situ tether formation. Progress also continues toward more complex reactions, including hydrolyses of b-amino nitriles and amides. This grant also opened a broad area for further exploration and applications: the use of aldehydes as cross-linking agents for organic molecules.









