Reports: DNI652157-DNI6: Non-adiabatic Dynamics of Small Polyphenyls Studied with Time-Resolved Spectroscopies
 printer friendly
printer friendlyWork in the last year has advanced our understanding of the non-adiabatic photochemical dynamics of ortho-arenes after UV photoexcitation. Since the early 1970's it has been known that photoinduced electrocyclization must be the first step in the light-induced synthesis of polyaromatic hydrocarbons from ortho-arenes (e.g. triphenylene from ortho-terphenyl, or OTP), but there have been no systematic studies of how structure-dynamics relationships dictate why these reactions have very high yields with some reactants and not others. This has changed the nature of the questions we seek to answer from on-going research: Specifically, we seek to determine how molecular structure influences the potential-energy landscapes and electronic state crossings that enable carbon-carbon bond formation via non-adiabatic photocyclization (Fig. 1). Not only does this work provide transformative experimental detail on structure-dynamics relationships that determine the likelihood of non-adiabatic processes, it is also relevant for establishing photochemical strategies for making polyaromatic networks that have potential material uses.
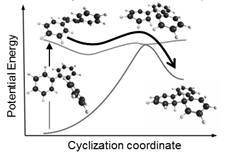
Figure 1: Representation of non-adiabatic photocyclization of ortho-terphenyl.
Transient absorption spectra measured at ultrafast delays following UV photoexcitation of OTP reflects an ultrafast photochemical reaction (Fig. 2a&b): Absorption bands in both the visible and UV regions appear immediately after photoexcitation and can be assigned to excited-state transitions of the reactant. These bands decay quickly after excitation and give rise to weaker features, one of which exhibits spectral shifting on a ~20 ps timescale that is consistent with relaxation of a vibrationally excited photoproduct created through an ultrafast process. Photoproduct absorption features are remarkably similar to those observed in the spectrum of cryogenically trapped dihydrophenanthrene, or DHP (Fig. 2, (c)); DFT calculations of the lowest-energy transitions for DHP and dihydrotriphenylene (DHT) explain this similarity and provides support to our assignment that non-adiabatic cyclization of OTP to form DHT occurs on a timescale of a few picoseconds (2.9 ps in tetrahydrofuran).
To understand the influence of molecular structure on the course of non-adiabatic photocyclization, we have investigated the photo-induced dynamics of 1,2-diphenylcyclohexene (DPCH). Excited-state spectra of DPCH exhibit remarkable similarity with both cis-stilbene and OTP and interconvert quickly to known absorption features of the anticipated photoproduct, a DHP analog (Fig. 3 top left). DPCH relaxes nearly ~2 times faster than OTP (Fig. 3 top right), which we interpret as the result of differences in structural constraints that influence passage to a state crossing (or conical intersection, CI) for cyclization. We therefore undertook a set of quantum chemical (CAS-SCF) calculations of the S0 and S1 minima and lowest-energy conical CIs in both molecules in order to understand how differences in molecular structure influence the excited-state potential-energy landscapes of these molecules (and, most likely, their dynamics). Calculated geometries of the two molecules exhibit differences along two key coordinates (Fig.3, bottom): Dihedrals spanning all three phenyl rings, θ, which corresponds primarily with twisting of the central C-C bond that bridges the terminal rings (so-called “ethylenic twisting” in diarylethylenes); and dihedrals between adjacent phenyl rings, φ, which corresponds primarily with torsional twisting of the terminal phenyl rings. Our calculations revealed that opening θ between the S1 minimum and the CI comes at a higher energetic penalty in OTP relative to DPCH for all active spaces used. This trend is consistent with the anticipated energetic penalty for straining the nominal sp2 hybridization of the bridging carbon atoms in the central ring of OTP. In contrast, the cyclohexene ring in DPCH can more readily accommodate strain introduced by twisting the ethylenic bond. The smaller energetic penalty for reaching the CI from the S1 minimum in DPCH is consistent with faster dynamics relative to OTP. This comparison of excited-state dynamics and structure was published in Journal of Physical Chemistry Letters and was our first publication on this topic.
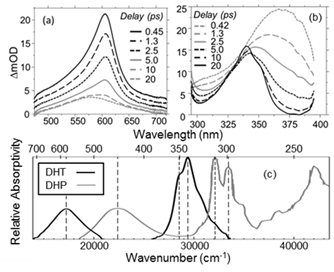
Figure 2: Ultrafast transient absorption spectra of photoexcited OTP collected at visible (a) and UV (b) probe wavelengths. (c) Spectrum of the photocyclized intermediate (dihydrotriphenylene, DHT) and dihydrophenanthrene (DHP).
Further work with OTP has examined the solvent dependence of OTP photocyclization in a collection of solvents with the goal of determining how the nature of solvent-solute interactions influences the shape of OTP's potential-energy surfaces and/or the dynamics that occur along them. Measurements reveal weak correlations between cyclization rate and solvent polarity or dielectric strength, suggesting that regions of the potential-energy surfaces relevant to OTP cyclization are weakly affected by static or dynamic solvation. In contrast, negative correlations with solvent viscosity and density indicate that solvent influences on excited-state dynamics occur primarily through mechanical interactions (i.e. friction). Although it is known that solvent friction at the molecular level need not correlate linearly with bulk solvent properties, these findings indicate that non-adiabatic photocyclization of OTP and its analogs are ideal processes for examining mechanical solvent influences on fast, low-barrier chemical dynamics. A publication describing these findings and the complete characterization of OTP photochemistry is currently in preparation.
Research activities described have greatly impacted the professional development of the graduate students involved due to the breadth of their experiences in the laboratory (ranging from spectroscopic measurements, theoretical calculations, and synthetic preparations). Furthermore, one student had the opportunity to present findings at the Gordon Research Conference on Photochemistry in July of 2013. Due to a time-sensitive availability of university start-up funds, only a small portion of funds from this grant were utilized in the last year. However, the remainder of funds will be required to continue supporting these students in the coming year. The PI's career has also greatly benefited from activities associated with this project: He has presented findings at two ACS meetings in the last year, raising his visibility in the fields of chemical dynamics and photochemistry. Importantly, progress in this research has led to new directions from the original proposal and the PI has been developing proposals for submission to other funding programs to garner support for continuing research beyond September 2014.
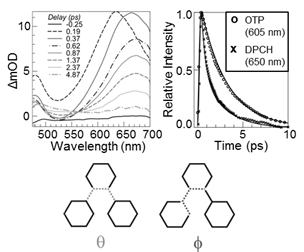
Figure 3: (Top left) TA spectra of UV-excited DPCH. (Top right) OTP vs. DPCH excited-state decay. (Bottom) Dynamics along these dihedral coordinates are critical to reaching the conical intersection for cyclization.









