Reports: ND251786-ND2: Site-Specific Isotope Fractionation of Hydrocarbons by Quantitative NMR Spectroscopy
 printer friendly
printer friendlyOverview
Although there is general agreement that oil and gas are produced through high temperature thermal cracking of sedimentary organic compounds, their operational modes still remain controversial. Bulk and compound-specific isotope analysis, and empirical and theoretical kinetic models of stable isotopes have made significant contributions to our knowledge on the sources and alteration pathways of oil and gases. However, their applications are still limited due largely to post-generative processes and a lack of suitable calibration data for kinetic modeling. Many organic molecules contain hydrogen and carbon in energetically nonequivalent positions (e.g., methyl and methylene groups of propane), and their isotopic composition can differ due either to equilibrium or to path-dependent kinetic processes. This site-specific or intramolecular 2H/1H and 13C/12C isotope fractionation between different H and C positions within a given hydrocarbon molecule (isotopomers) can provide additional information and much deeper insights into their sources and alteration pathways of petroleum hydrocarbons within source and reservoir rocks, including temperatures, conditions, and pathways of thermal cracking and other formation processes. In this proof of principle study, we propose to demonstrate that site-specific 2H/1H and 13C/12C isotope fractionations can be determined with sufficient precision for select light hydrocarbons and related petroleum products by means of quantitative 2H and 13C NMR spectroscopy.
Analytical Methods
The focus of the research is on the light alkanes, the major components of petroleum fluids. Initial analysis consisted of 2H NMR spectroscopy of cyclohexene and pentane. These were chosen because of the simplicity of the compound and the resulting NMR spectrum, and deuterium in differing positions on the molecule (aromatic, CH2, CH3) that would result in both easily separable resonances and with differing site specific isotopic ratio.
1H, 2H, and 13C NMR spectra were recorded on a JEOL SCC-400MHz spectrometer operating at 399.7822 MHz (1H), 61.369 MHz (2H), 100.5253 MHz (13C). Correct assignment of resonances was confirmed by performing 1H, and 2-dimensional NMR spectroscopy in chloroform. The instrument was automatically tuned for the appropriate nucleus prior to analysis.
Deuterium was tuned on the low frequency channel, as it is more sensitive than the lock channel. Samples for deuterium analysis were run neat and without lock in a Wilmad 5 mm Step Down Ultra-Thin Wall Precision NMR Sample Tube. Proton decoupling was used to eliminate splitting of the resonances. Acquisition time was sufficiently long to allow for the full decay of the FID, and a relaxation delay was greater than or equal to five time T1 allowing for the nucleus to fully relax. The number of scans was chosen such that the smallest resonance had a S/N of 150 or greater. The S/N was improved with the line broadening factor, sexp = 0.2. The digital resolution was increased by zerofilling 4 times. Phasing was performed automatically but manually refined. The resolution, checked by measuring the peak width at half height without line broadening, was less than 0.5 Hz.
Tetramethylurea (TMU) was purchased from the European Commission Joint Research Centre Institute for Reference Materials and Measurements (IRMM) and is used as an 1H/2H isotopic standard. In a sealed J-Young style NMR tube, TMU was mixed with the analyte to a concentration that would yield the TMU peak at the same integration as the smallest analyte peak.
Results and Discussion
The NMR spectrum for pentane (Figure 1) shows three peaks separable in the deuterium spectrum. The statistical distribution of deuterium is in a 4 : 2 : 6 ratio. The observed ratio, 4 : 2.0126 : 5.7163, differs from the statistical distribution in the CH3 position. This was confirmed with 10 trials, yielding a standard deviation of the CH3 position of 0.06269 (1.1%). Whereas compounds resulting from biological processes (ethanol, vanalin, etc.) have shown a variation in their 2H fractionation ratios as much as 300‰ or greater, the pentane analyzed in this study has a variation of 5.2% in the CH3 site compared to the CH2 sites.
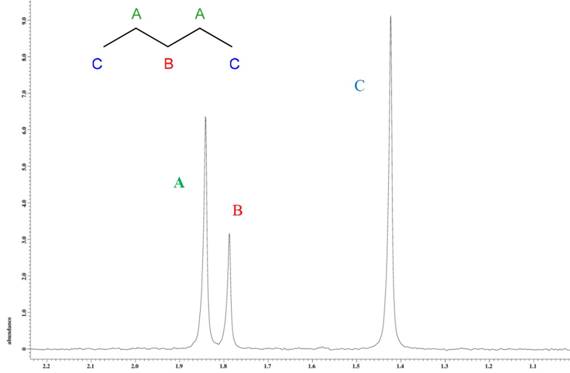
Figure 1. 2H NMR spectrum of neat pentane.
The NMR spectrum of cyclohexene (Figure 2) shows three resonances, one alkene and two alkane, in the deuterium spectrum. The statistical distribution is expected to be 2 : 4 : 4, but the observed ratio is biased toward the alkene at 2 : 3.852 : 3.8318. This was also confirmed with 10 trials, yielding a standard deviation at the CH2 positions of 0.053556 and 0.050789 (both 1.3%). Cyclohexane has a variation of 3.8% in the CH2 site compared to the alkene site.
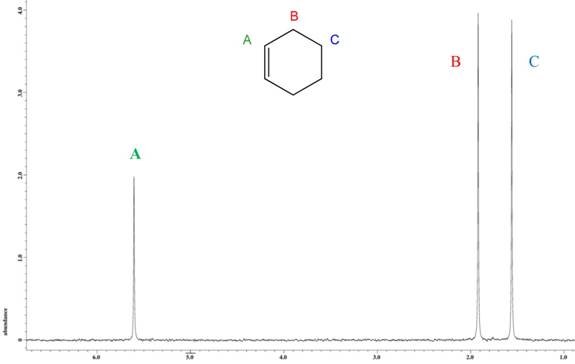
Figure 2. 2H NMR spectrum of neat cyclohexene.
Comparison of integrations only affords a ratio of isotope fractionation within a given molecule. To measure deuterium contents, an international standard, TMU, is utilized along with the analyte (Figure 3). The standard is certified by IRMM with a deuterium-to-hydrogen ratio of 141.9 × 10-6, and is used to determine the deuterium-to-hydrogen ratio at each site. At position A, the ratio is 132.9×10-6, position B is 124.4×10-6, and position C is 127.5×10-6.
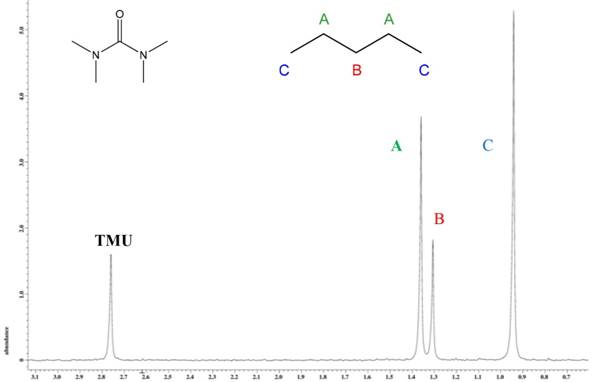
Figure 3. 2H NMR spectrum of TMU and pentane.
Conclusion and Future Work
Deuterium analysis of the small hydrocarbons, pentane and cyclohexene, demonstrate the feasibility of site-specific 2H/1H isotope fractionation, which corresponds to the type of chemical environment.
Work is ongoing to determine the fractionation in other small organic molecules such as butane, p-xylene, and others (Figure 4). All of the compounds afford two or more unique sites resolvable by proton decoupled deuterium NMR spectroscopy. Site-specific 13C/12C isotope measurements of the same compound will also be initiated.
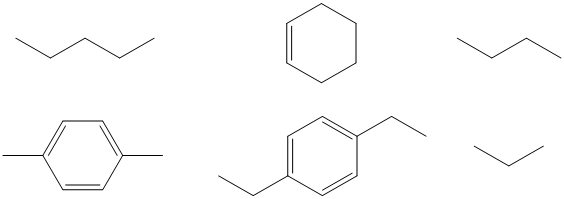
Figure 4. Target molecules for determination of site-specific 2H/1H isotope fractionation.
Funding from NSF grant CHE-1048553 and the NSF's CRIF program which supported the purchase of this instrument, is gratefully acknowledged.









