Reports: DNI651000-DNI6: Effects of Quantum Motion of Nuclei on Reactivity of Hydrocarbons Studied in the Approximate Quantum Trajectory Framework
 printer friendly
printer friendlyResearch in our group is focused on studies of quantum-mechanical (QM) behavior of nuclei and its effects on reactivity of molecular systems in gas and condensed phases. The dominant quantum-mechanical effects, present even in large molecular systems, are tunneling, internal energy flow and nonadiabatic dynamics involving excited electronic states. In large molecular systems, however, one expects that not all nuclei exhibit quantum effects, but just a few selected degrees of freedom describing, for example, proton motion leading to tunneling at low energies. The zero-point energy (ZPE) of the vibrational degrees of freedom may affect the energy flow even for heavier particles. Inclusion of the ZPE into (quasi)classical trajectory simulations is an outstanding conceptual problem. Thus, a rigorous yet practical hybrid quantum/classical dynamics approach is a long-standing goal of theoretical chemistry. We are using the quantum trajectory (QT) formulation of the Schrödinger equation to define and analyze quantum/classical or, more precisely approximate-quantum/nearly-classical dynamics. The approximate Mixed Quantum/Classical Trajectory (MQCT) dynamics developed in our group is used for conceptual and for system-oriented studies, such as isotope effects in proton transfer and adsorption in systems of 10-100 atoms. Within the quantum trajectory formalism the approximations -- necessary for applications to large systems -- are driven by physical considerations and well-identifiable. The approach is particularly appealing since it treats all nuclear degrees of freedom on equal footing and is readily compatible with electronic structure solved on-the-fly.
The specific research goal supported by this grant is a study of quantum effects in systems consisting of carbon and hydrogen atoms. To test the MQCT formalism we modeled nonreactive collision of He (classical degree of freedom, y) with HBr (quantum degree of freedom, x). The quantum correction is a function of the coordinate x, but parametrically depends on the domains defined in the y-coordinate. The conceptual challenge is description of the energy transfer between these two degrees of freedom, without losing the zero-point energy of the quantum degree of freedom to the classical degree of freedom. Figure 1 illustrates the model system (left panel) and application of MQCT (central and right panels).
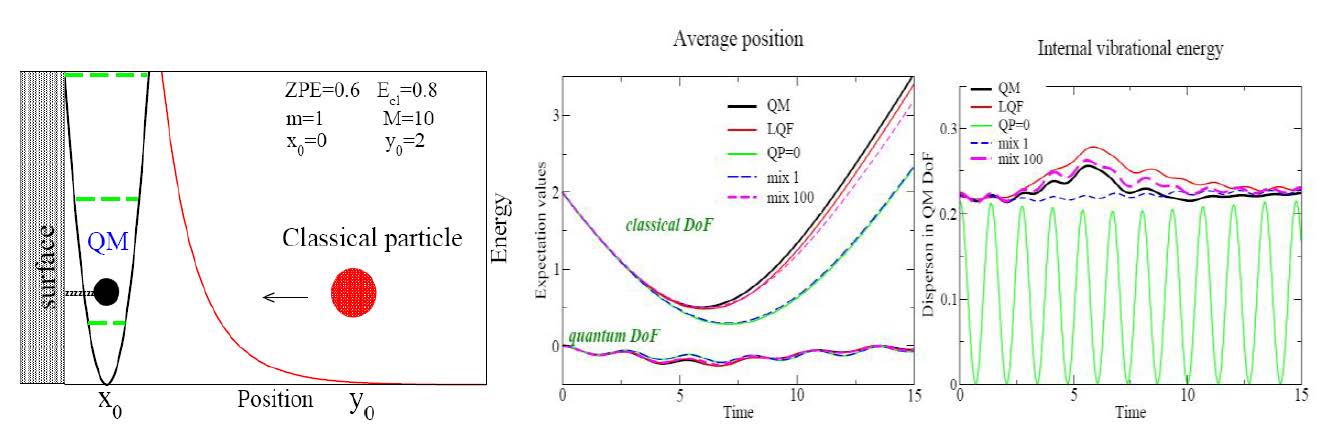
Figure 1. Left panel: He+HBr model system. Average positions (central panel) and vibrational energy of HBr (right panel) as functions of time, obtained with exact quantum-mechanical propagation (black), approximate quantum trajectory propagation in two dimensions (red), without the quantum correction (green), Ehrenfest (blue dash) and MQCB (magenta).
Next, the MQCT approach has been applied to explore nuclear quantum effects on adsorption of hydrogen on a graphene flake C37H15 (also C87H23). The dominant quantum effect is due to localization of the initial proton wavefunction affecting dynamics of the “flake” depending if the quantum correction for the proton is included (labeled “LQF” on graphs) or not (“classical”).
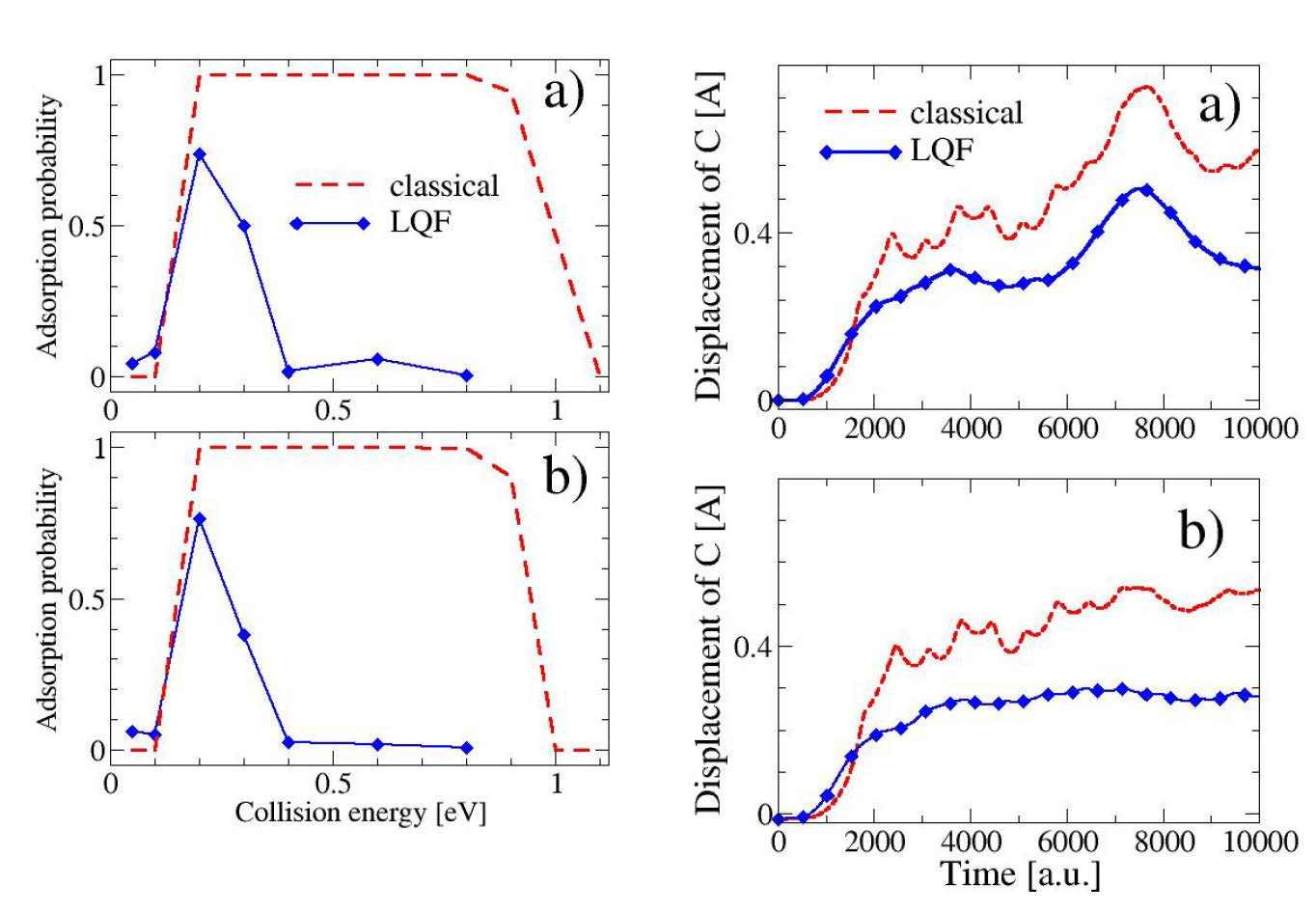
Figure 2. H colliding with C37H15 at energies 0.1-1 eV. Quantum proton is represented with an ensemble of quantum trajectories (white spheres above the graphene flake) . Vibrations of the flake dissipate collision energy and are necessary for hydrogen adsorption. The wavepacket was represented with 2400 quantum trajectories that were propagated for 1000 time steps with the classical forces coming from the Density Functional Tight Binding method.
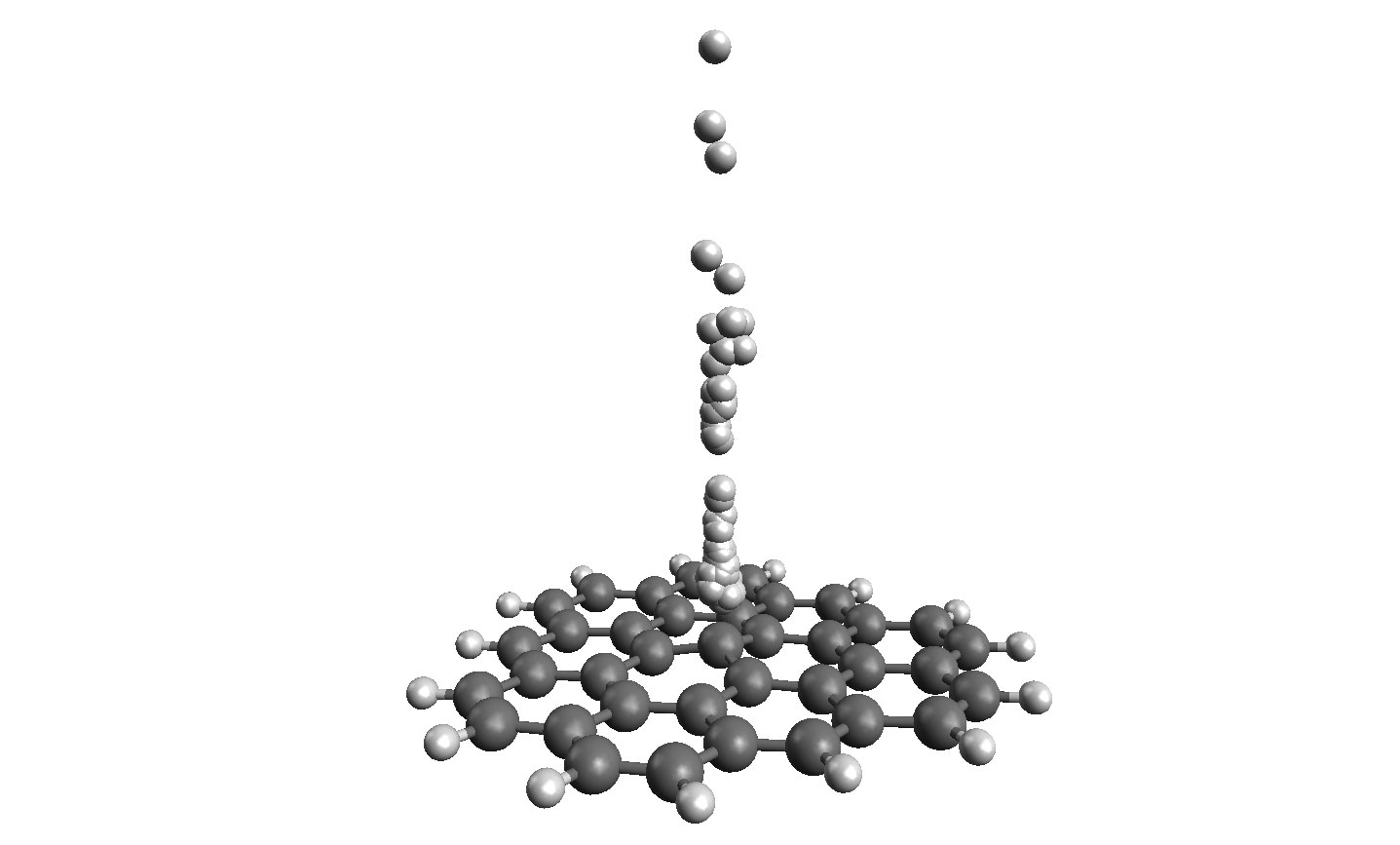
Figure 3. Adsorption probability (left) as a function of collision energy and displacement of the central carbon atom (right) as a function of time for C37H15 (a) and C87H23 (b).
The paper “A Hybrid Quantum Trajectory-Electronic Structure Approach for Exploring Nuclear Effects in the Dynamics of Nanomaterials” S. Garashchuk, J. Jakowski, L. Wang, B.G. Sumpter is in review in the Journal of Chemical Theory and Computation. Current work includes analysis of scattering off various sited on the graphene flake; future work will focus on a more challenging scattering of molecular hydrogen.
The grant has been also used to support development of the analytical triplet and singlet surfaces for O+C2H2 (diagram below) reaction carried out in conjunction with experimental work from the Minton group from Montana State University. Extensive benchmarking of the surface against electronic structure reaction path calculations for the major channels have been performed. The surfaces are described, analyzed and prepared for distribution to researchers through Chemical Physics Letters. The manuscript “Analytical potential energy surface for O+C2H2 system” by S. Garashchuk, V. A. Rassolov and B. J. Braams is in review.
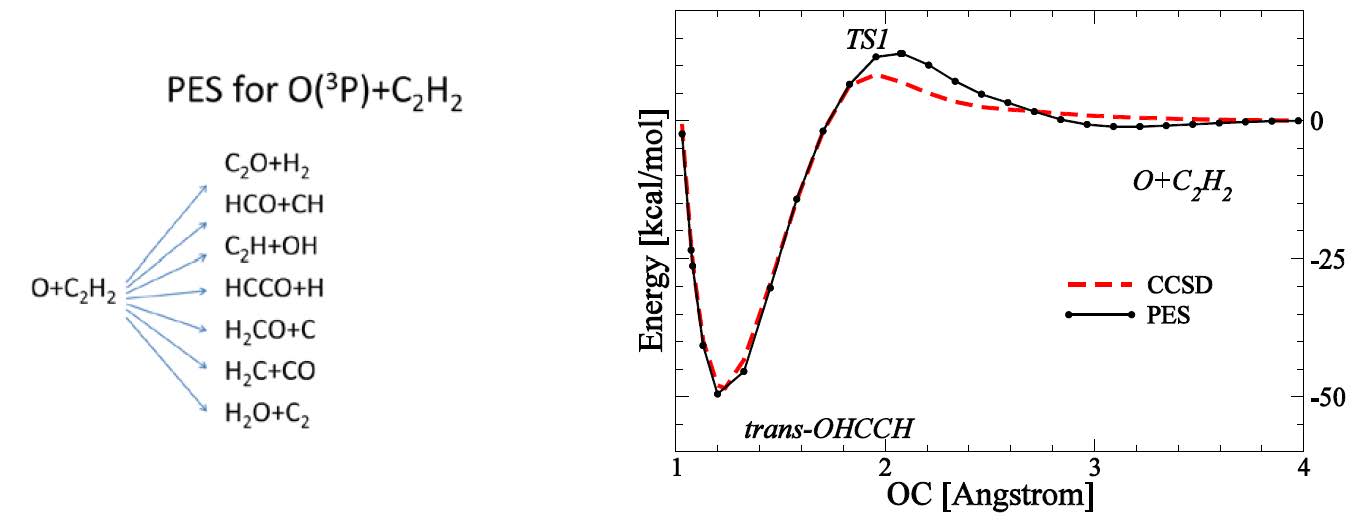
Figure 4. Reaction channels included into the triplet surface (left) and a representative reaction path obtained from the developed surface and coupled cluster calculation (for geometries optimized using Density Functional Theory).









