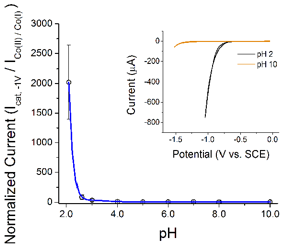
Reports: UNI551724-UNI5: Surface Immobilization of Cobalt-Diglyoxime Complexes: The Effects of Interfacial Chemistry on Electrocatalytic Activity in Aqueous Media
 printer friendly
printer friendly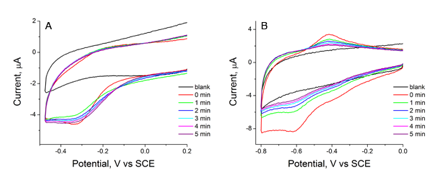
Figure 1: Cyclic voltammograms (100 mV/s) of a Co(dpgBF2)2 / pyridine modified glassy carbon electrode recorded in (a) dichloromethane (0.1 M TBAPF6) or (b) water (0.1 M sodium phosphate, pH 7.0). The cobalt signal was stable in dichloromethane but not in water.
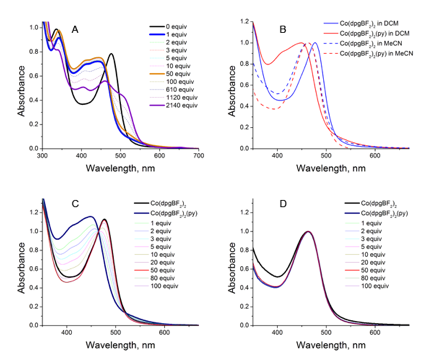
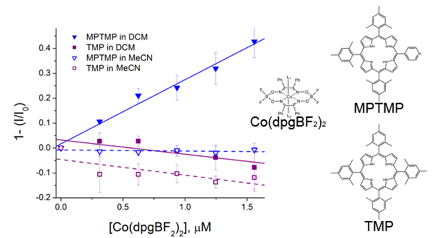
Figure 3: A Stern-Volmer plot showing quenching of the porphyrin singlet-excited state as a function of Co(dpgBF2)2 concentration for MPTMP and TMP in dichloromethane and acetonitrile. Quenching was only observed for the pyridyl porphyrin in dichloromethane.