Reports: DNI352325-DNI3: Cation-Crown Ether Interactions to Control Ligand Hemilability and Catalyst Function
 printer friendly
printer friendlyIntroduction. Hemilabile ligands play a crucial role in many metal-catalyzed processes through reversible binding of the metal center. When one arm of a chelate dissociates, substrate can bind the metal center; after turnover, re-association can stabilize the coordinatively unsaturated metal center. While ligand hemilability is recognized as an important catalyst trait, designing catalysts that feature hemilabile ligands is difficult. If a ligand does not have the correct binding dynamics, catalysis may slow down or stop entirely (e.g., no dissociation), or the catalyst may decompose (e.g., full dissociation). Tuning the properties of the hemilabile ligand normally requires the potentially onerous synthesis of a new ligand with different steric or electronic properties. The goal of this ACS PRF New Doctoral Investigator project is to synthesize a new class of "pincer-crown-ether" ligands that combine the strong binding ability of pincer ligands with an aza-crown ether group that can modulate hemilability. Through the macrocyclic receptor site, this new ligand framework features cation-triggered hemilability, as the crown-ether donors bind a cation additive instead of the transition metal center. Cations display a range of binding affinities to crown ether macrocycles, which allows facile tuning of the hemilabile activity.
Results and discussion. In the first year of funding, we have successfully synthesized new phosphino-aza-crown ether ligands and found that these ligands exhibit flexible coordination chemistry. Working with a first year graduate student, the ligands were accessed by a three-step synthesis from commercially available 3-hydroxymethylphenol. The concise synthesis facilitates access to diverse ligand structures. Attempts to metallate the tert-butyl-phosphine derivative [tBuPOCN15c5]H were thwarted, perhaps owing to the demanding steric bulk of the tert-butyl substituents. Accordingly, the iso-propyl-phosphine derivative [iPrPOCN15c5]H was synthesized and reacted smoothly when treated with Ir(CO)2(Cl)(toluidine) to form carbonyl hydrochloride 1 (Scheme 1). Multinuclear NMR studies suggest that complex 1 adopts the expected tridentate (PCN) binding mode to give an octahedral complex. A five-coordinate species was targeted by reaction of [iPrPOCN15c5]H with [Ir(COD)Cl]2, which cleanly yielded hydrochloride 1 (Scheme 1). An X-ray diffraction study performed on 1 revealed the unexpected coordination of an ether group of the crown ether macrocycle, rendering the ligand tetradentate (k4), as shown in Figure 1. Upon treatment of 1 with NaBArF4 (ArF is 3,5-bis(trifluoromethyl)phenyl), chloride abstraction resulted in clean formation of [(k5-POCN15c5)Ir(H)][BArF4] (2). An X-Ray diffraction study revealed pentadentate (k5) binding of the pincer-crown-ether ligand (Figure 1). These studies reveal how an unexpectedly flexible ligand can adaptively bind the metal to stabilize low-coordinate species, readily sampling three different coordination numbers.
With a series of complexes featuring different pincer coordination numbers in hand, we began initial investigations into binding dynamics. Treatment with Lewis bases suggests that the macrocyclic ether ligands are weakly bound: Treatment of 2 with ~30 equiv MeCN cleanly produced a new product, identified as (k3-POCN15c5)Ir(H)(Cl)(NCMe). Similarly, treatment of pentadentate complex 3 with MeCN readily produced [(k3-POCN15c5)Ir(H)(NCMe)2]+. Both reactions are reversible, with solvent evaporation restoring the iridium-ether interactions. The bond strength is also being investigated computationally.
Judging the Ir–O bonds to be sufficiently weak, we undertook reactions in the presence of cationic alkali metal Lewis acids. Addition of LiOTf to complex 3 facilitates ligand binding, as THF and MeCN bind Ir much more readily than in the absence of LiOTf. Furthermore, after treatment of pentadentate complex 3 with LiOTf in the presence of MeCN, the reaction was no longer reversible: removal of solvents afforded [(Li@k3-POCN15c5)Ir(H)(MeCN)2]+ (the @ symbol denotes Li+ intercalation in the macrocycle).
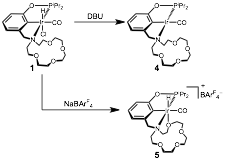
Based on our interest in promoting carbonylation chemistry with the Lewis acidic sites, we have also been seeking appropriate precursors for this chemistry. A variety of carbonyl derivatives have been synthesized, as outlined in Scheme 2. Hydrido carbonyl 1 can be treated with base to afford Ir(I) carbonyl complex 4. Alternatively, chloride abstraction from 1 generates cationic [(iPrPOCN15c5)Ir(H)(CO)]+ (5), which likely adopts a k4 pincer ligand geometry. These species are currently being investigated for C–H activation and C–C bond formation reactivity.
Summary. Preliminary results suggest that pincer-crown-ether ligands can support rich, flexible coordination chemistry. Initial reactivity with alkali metal cations suggests that macrocycle bonds to the metal center can be broken upon addition of cationic additives. The ACS PRF DNI funding has supported a talented first year graduate student. The work was recently presented at the 245th ACS National Meeting and at the Organometallic Chemistry Gordon Research Conference, and manuscript will be submitted soon. Through generous support from the ACS, the modular pincer-crown-ether ligand framework has developed into a strong basis for research in our group, rapidly diversifying into an array of projects in organometallic catalysis.
Scheme 1
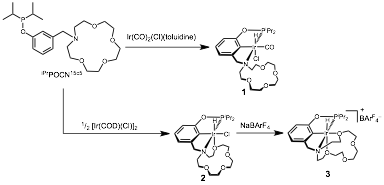
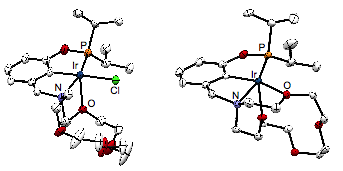
Figure 1. Structural representations of 2 and 3, with ellipsoids at 50% probability. Hydrogen atoms, solvent molecules and counter ions omitted for clarity.
Scheme 2