Reports: DNI151707-DNI1: Novel Catalytic Enantioselective [3,3]-Sigmatropic Rearrangements for the Preparation of Highly Substituted Fused Heterocycles and Biaryls
 printer friendly
printer friendlyThe Kürti group is pursuing the development of powerful and practical new methods for the synthesis of highly functionalized symmetrical and unsymmetrical biaryls. Ideally these new routes are expected to: (a) be operationally simple; (b) utilize readily available and inexpensive starting materials; (c) avoid the use of TM catalysts, ligands and strong oxidizing agents; (d) achieve C-C bond-formation with controllable and high regioselectivity; (e) build molecular complexity rapidly and with exceptional step-economy (i.e., in one-pot); (f) permit the presence of functional groups that are generally not compatible with TMs (i.e., halogens, that allow further functionalization via the cross-coupling manifold) and (g) be complementary to existing cross-coupling methods by allowing the facile preparation of currently unknown or otherwise inaccessible biaryls. Given the remarkable track record of axially chiral biaryls as highly efficient ligands in asymmetric synthesis, it is clear that convenient synthetic access to many structurally diverse and uniquely functionalized biaryls will have a profound impact on the discovery of new chemical reactivity.
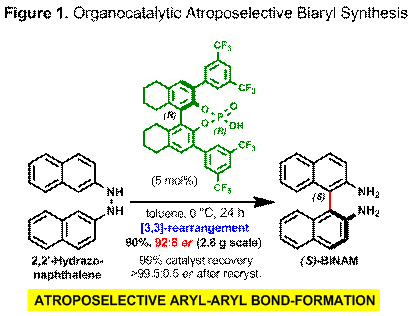
We have successfully developed an organocatalytic aryl-aryl bond-forming process for the regio- and atroposelective synthesis of 2,2'-diamino-1,1'-binaphthalenes (BINAMs). In the presence of catalytic amounts of axially chiral phosphoric acids, achiral N,N'-binaphthyl hydrazines undergo a facile [3,3]-sigmatropic rearrangement to afford enantiomerically enriched BINAM derivatives in good to excellent yield (Figure 1). This transformation represents the first example of a metal-free, catalytic C(sp2)-C(sp2) bond-formation between two aromatic rings with concomitant de novo atroposelective installation of an axis of chirality. Density functional calculations reveal that in the transition state for C-C bond formation, the phosphoric acid proton of the catalyst is fully transferred to one of the nitrogen atoms of the substrate and the resulting phosphate acts as a chiral counterion. We can predict the absolute configuration of the axially chiral biaryl products. This approach also allows us to engage in the rational design of more effective catalysts and explore related rearrangements. We are in the process of expanding the scope of this powerful atroposelective method to unsymmetrical biaryl hydrazines. The axially chiral and structurally diverse biaryl products are expected to find broad utility in asymmetric catalysis, drug discovery and materials science.
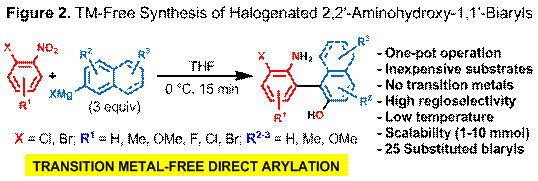
We have also successfully developed an operationally simple, transition metal (TM)-free, regioselective and direct aryl-aryl bond-forming process for the synthesis of halogenated 2,2'-aminohydroxy-1,1'-biaryls that are currently either inaccessible or challenging to prepare in an atom- and step-efficient manner using conventional methods (Figure 2). The addition of an arylmagnesium halide to an ortho-halogen-substituted nitrobenzene at low temperature generates a transient N,O-biarylhydroxylamine, which rapidly undergoes either [3,3]- or [5,5]-sigmatropic rearrangement to form, in one-pot, the corresponding 2-amino-2'-hydroxy-1,1'-biaryl or 1-amino-1'-hydroxy-4,4'-biaryl, respectively. Peri-selectivity is controlled by the choice of solvent as well as temperature. This direct arylation process is also readily scalable (1-10 mmol). Density functional calculations suggest that from the N,O-biarylhydroxylamine intermediate there is a low energy stepwise pathway that involves initial Mg-mediated N-O bond cleavage followed by pathway branching toward either [3,3]- or [5,5]-rearrangement products involving C-C bond formation and finally re-aromatization.
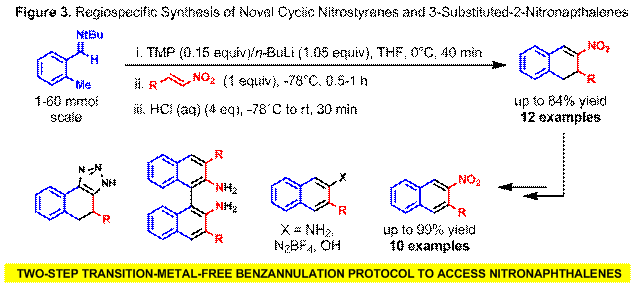
To support the synthesis of structurally complex BINAM derivatives, we have developed a two-step, practical, regiospecific and readily scalable benzannulation protocol for the preparation of novel 3-alkyl/aryl substituted 2-nitronaphthalenes (Figure 3). Addition of a b-nitrostyrene or nitroalkene to a solution of freshly prepared lithiated o-tolualdehyde tert-butyl imine first leads to the formation of a nitronate, via rapid 1,4-addition, then an intramolecular aza-Henry reaction takes place to afford a six-membered carbocycle. Subsequent treatment of the reaction mixture with aqueous acid affords novel substituted cyclic nitrostyrenes that can be conveniently aromatized via a one-pot radically-induced bromination/elimination sequence to furnish the corresponding 3-alkyl/aryl-2-nitronaphthalenes in excellent yields. This facile two-step benzannulation may serve as a prototype for related transformations that build molecular complexity rapidly and with exceptional step-economy. Moreover, the synthetic community now has access to 2-aminonaphthalene building blocks for the preparation of currently unknown symmetrical and unsymmetrical BINAM derivatives that will serve as ligands or catalysts in a variety of asymmetric transformations. Toward this end, the straightforward synthesis of 2-aminonaphthalenes, substituted BINAMs, 2-naphthols as well as tricyclic fused 1,2,3-triazoles have also been achieved.
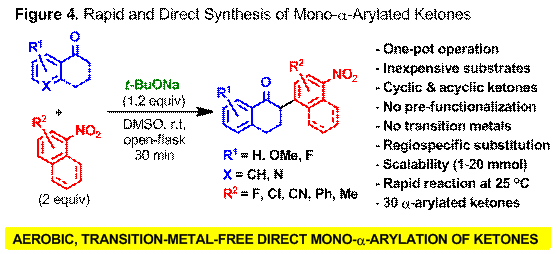
We have demonstrated that mono-a-arylation of ketones is preparatively feasible under aerobic and TM-free conditions (i.e., green chemistry) to yield products that feature substitution patterns that are either very challenging or impossible to obtain using conventional methods (Figure 4). The transformation is operationally simple, scalable, the scope of substrates is wide and there is no need for pre-functionalization (i.e., a-halogenation or silyl enol ether-formation) or the use of specialized arylating agents (i.e., diaryliodonium salts). DFT calculations suggest that the in situ-generated enolate undergoes direct C-C bond formation with the nitroarene followed by regioselective O2 mediated C-H oxidation. Studies to expand this direct a-arylation process to other classes of carbonyl compounds and electron-deficient arenes are currently under way in our laboratories.