Reports: DNI1052138-DNI10: Toward Efficient Design of Noncentrosymmetric Oxyfluorides: Ab Initio Crystal Engineering of Next-Generation Catalysts
 printer friendly
printer friendlyIntroduction: Materials exhibiting noncentrosymmetric (NCS) crystal structures lack inversion symmetry. They are important for stimulating efficient and high-yield asymmetric catalysis of fossil resources (petroleum, coal, and natural) into consumable fuels for transportation, industrial production and electricity generation. Despite their industrial functionality, efforts to design and discover new NCS materials remains challenging, due in part, to the limited control over the fundamental anion building units coordinating cations with chemistries susceptible to NCS displacements. To overcome these materials disparities, this project explores the design of crystal structures in cryolite and elpasolite-structured mixed anion (oxyfluoride compounds). The goal is to build a reliable set of crystal-chemistry guidelines that dictate the optimal geometries and chemistries favorable to NCS structures. That knowledge will then be harnessed to computationally design promising NCS oxyfluorides. By doing so, we will identify new classes of support materials for catalytic processing of synthetic fuels.
Results: The oxyflouride ligand combination is a promising anion network to exploit for NCS materials formation, because the interactions among the more (M–O) and less (M–F) covalent bonds are conducive to stabilizing cooperative NCS distortions. We began our study of these interactions in mixed anion system by first investigating the fully fluorinated compounds Na3MnF6 and Na3ScF6 to build an understanding of the atomic distortions in cryolite compounds. Although neither material is NCS, Na3MnF6 is of particular interest, because it of its first-order Jahn-Teller Mn3+ activity and experimental reports that it undergoes a structural phase transition at moderate hydrostatic pressure (~2 GPa). To understand the origin of this transition, we carried out electronic structure calculations based on density functional theory within the general gradient approximation (GGA) of Perdew-Burke-Ernzerhof revised for solids (PBEsol). As a starting point we computed the zero-pressure structures. Our calculations for these structures provide good agreement with the experimentally reported equilibrium phases.
We then computationally explored the phase stability of the compound as a function of hydrostatic pressure (Figure 1). We found that a structural transition occurs via a reorientation of the d(z2-r2) Jahn-Teller axis from along the crystal c-axis at low pressure to within the ab-plane near 2.15 GPa. In contrast, the d0 Sc compound has the same low-symmetry structure (P21/n) as Na3MnF6, yet the absence of any pressure-induced transition and Jahn-Teller distortion in this system corroborates our finding that the Mn d4 electronic configuration is crucial. Further insight into the nature of the transition was provided by group theoretical analysis of the atomic structure. Investigating the irreducible representations (irreps) of the low and high-pressure phases with reference to the aristotype reveals that the Jahn-Teller mode in concert with the zone boundary mode are responsible for the reorientation of the d(z2-r2) orbital and Jahn-Teller axis at high pressure., We also examined several magnetic states since we are unaware of any experimental reports detailing the electronic and magnetic properties of Na3MnF6. We predict the fluoromanganate to be a ferromagnetic insulator.
Figure 1: Evolution of the total energy for the low pressure, P21/n(1) phase and the high pressure, P21/n(2) phase with cell volume. The zero-pressure structure with disconnected MnF6 and the Na cation lattice are highlighted in the inset.
|

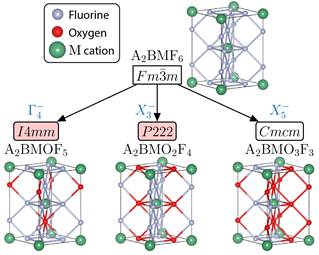
In the pursuit of the design new NCS oxyfluorides, we next attempted to exploit the inherent acentric character of transition metal (M) building units MOxF6-x in elpasolite-structued compounds (A2BMX6, X=MOxF6-x). Our electronic structure calculations, however, require as input the crystal structure. In order to systematically investigate the structural phase space enabled by mixed-anion ordering, we formulated a group-theoretical protocol to enumerate derivative structures (children) accessible from the aristotype Fm-3m parent phase. In our mode crystallographic study, we imposed irreps of the parent phase that split the occupancy of the anion Wyckoff positions while also lowering the symmetry under the constraint that required stoichiometry, i.e., MOxF6-x, (x = 1, 2, 3), is maintained. Since group theory exploits mathematical relationships without the a priori knowledge of chemistry we are able to probe all possible octahedral anion orderings. We then classified the structures into cis-, trans-, fac- and mer-orderings based on the arrangement of the fluorine and oxygen atoms. This approach has resulted in registry of over a hundred new structures (nonpolar, polar, and chiral) consistent with the oxyfluoride elpasolite chemistries (Figure 2). We will next explore the energetically stability of this phases in search of new NCS compounds.
Figure 2: Compositional irreps and structure relationships that give ligand ordering in elpasolites, which are drawn to emphasize the O and F anion sublattice around the M cation. (Shaded space group labels indicate NCS crystal classes; A and B atoms omitted.)
|
The financial support from the Petroleum Research fund has been critical to launching a materials discovery paradigm within my group. It has also led to a new understanding of pressure-induced phase transitions in fluorine compounds. In year 1, the project has funded 1 full-time PhD student; it has also formed the focus of one undergraduate senior-design project. Travel funds have enabled the PhD student to attend the annual March Meeting of the American Physical Society meeting, and also to participate in an international workshop on electronic structure methods aimed at training young scientists in computational methods. The latter is an important experience, which will have lasting impact on his future research career.









