Reports: UR150086-UR1: Oxidation of Alkenes in Aqueous Solvent Mixtures Using Environmentally Benign Reagents
 printer friendly
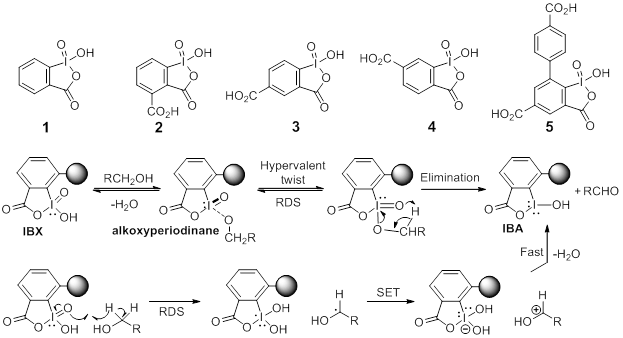
printer friendlyContinued efforts in our laboratory are aimed at the design, synthesis and reactivity of water-soluble derivatives of o-iodoxybenzoic acid (IBX, 1). Incorporation of a carboxylic acid functional group on the arene ring of IBX as in 2, 3 and 4 has been found to enhance the solubility of these reagents in water. Unique chemoselectivities observed during the oxidation of alcohols and other substrates using 2, 3 and 4 have prompted us to propose a reaction mechanism that involves a H-atom abstraction from the a carbon of the alcohol to initiate oxidation with a concomitant reduction of I(V)→I(IV) as shown in the bottom half of Scheme 1. The established mechanism for oxidation of alcohols using IBX in polar aprotic solvents involve the formation of the
Scheme 1: Structures of water-soluble IBX derivatives and mechanisms of alcohol oxidations
alkoxyperiodinane intermediate which undergoes a reductive elimination to provide the oxidized product along with IBA, the reduced form of IBX, as shown in the top half of the Scheme 1. Recently theorized (DFT calculations) hypervalentiodine twist notes that the disproportionation of the alkoxyperiodinane intermediate undergoes a coordinated motion of ligands around the iodine center so that the oxo ligand attains planarity and the transition state begins to resemble the planar product IBA. The presence of bulky groups at ortho position of the IBX ring is predicted to accelerate the twisting of the alkoxy ligand to relieve the repulsive steric interactions present in the initially formed alkoxyperiodinane adduct. The rate enhancement in the alcohol oxidation that can be attained by having suitably sized (not too large) ortho substituent will only be observed in polar aprotic solvents where the ligand exchange mechanism is in operation. We expect, as predicted by Su and Goddard,1 that the initial ligand exchange step will be disfavored with the use of reagent, 5, with a bulky phenyl substituent at the ortho position and thus result in substantially reduced rates of oxidation of alcohols. However, the presence of the bulky phenyl group in 5 should have no effect on the rate of oxidation carried out in aqueous solvent mixtures where we believe a hydrogen atom abstraction is the rate limiting step. The synthesis of 5, the progress to-date along with additional oxidative transformations uncovered during the past year is narrated in this annual report.
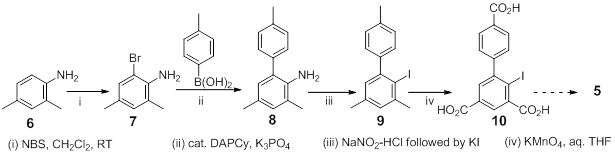
Bromination of 2,4-dimethylaniline, 6 with N-bromosuccinimide in dichloromethane gave 7 in 60-70% yield. An optimized Suzuki coupling procedure utilizing bis(dicyclohexylamine)palladium(II)-acetate (DAPCy) along with K3PO4 provided a modest
Scheme 2: Towards the synthesis of 5
40-50% yield of the couple product 8. Diazotization in aqueous ethanol followed by treatment with KI gave 9 which was subsequently oxidized in aq. THF using KMNO4 to yield the tri-acid 10, the immediate precursor to 5. The tedious isolation and purification of 10 has been successfully carried out using a soxhlet extraction procedure. We are currently accumulating sufficient quantities of 10 before an attempted oxidation to yield 5 will be attempted.
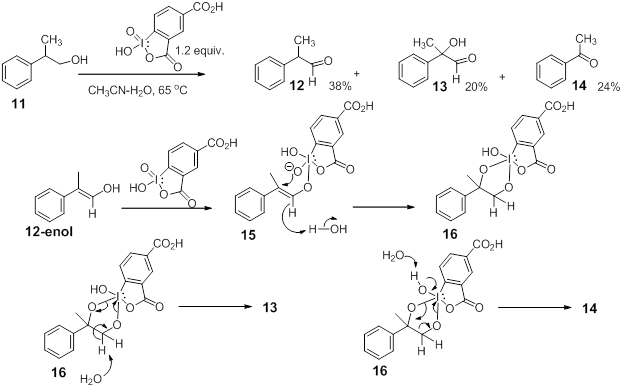
Though alcohol oxidation has remains the primary application of IBX, many novel oxidative transformations continue to be uncovered using IBX, with contributions from Nicolaou lab for benzylic C-H oxidation setting the stage in this arena.2 Last year while investigating the unusual oxidation behavior of 2-phenylethanol derivatives using water-soluble IBX derivatives we uncovered an unexpected C-C bond cleavage during the oxidation of 2-phenyl-1-propanol,11 to yield acetophenone 14. While the formation
Scheme 3: Plausible mechanistic insights into the formation of 13 and 14 from 11
of the hydroxyaldehyde, 13 through oxygen transfer from IBX was not an unexpected observation, the formation of acetophenone,14 continues to puzzle us and investigations to delineate plausible mechanisms are currently underway.3 Our preliminary hypothesis is that both 13 and 14 are formed from a common cyclic intermediate, possibly the cylic-diester intermediate, 16 formed as shown in Scheme 3. The two possible disproportionations of 16 can lead to the formation of 13 and 14 as shown. The postulated formation of 16 through 12-enol prompted us to look into the oxidation of b-dicarbonyl compounds with readily available enol-tautomers. It was hypothesized that oxidation of b-diketones and b-ketoesters with the water-soluble reagent 3 could lead to a-hydroxy-dicarbonyl derivatives along with oxidatively cleaved products. We have now embarked on a substrate scope study to probe the feasibility of the projected transformations. The initially chosen b-dicarbonyl substrates include

Scheme 4: Oxidation products of b-diketone and b-ketoester substrates
both b-diketones and b-ketoesters as shown in Scheme 4. The substrates were treated with 1.3 equiv. of 3 in aqueous acetonitrile (1:1 v/v) at 65°C for 6h. The reported yields of the hydroxylated products are isolated (column chromatography) yields. The presence of the dehydrogenated product and the oxidatively cleaved product were readily discernible from characteristic signals for the respective compounds in the HNMR spectra of the crude product mixtures. It is worth noting that while the dehydrogenated products are obtained in trace amounts from all of the substrates, the keto-ester 21 gave the dehydrogenated product as the major and readily isolable product in 80% yield. Our efforts in this area in the coming months will be directed at understanding whether it would be possible to tweak the reaction conditions and or substrate structures to control the product outcome from these oxidative transformations. We believe that developing a general synthetic methodology for any of the three product type would be a desirable synthetic goal.
In summary, we continue to make progress in many areas of the originally proposed research. Successful culmination of the two ongoing mini projects will result in peer-reviewed publications in the near future. Both undergraduate and graduate students (MS) continue to get trained in all aspects of chemical research through this funded proposal.
References:
- Su, J. T.; Goddard, W. A. J. Am. Chem. Soc. 2005, 127, 14146-14147.
- Nicolaou, K. C.; Bran, P. S. Zhong, Y.-L. J. Am. Chem. Soc. 2001, 123, 3183-3185.
- Thamisetti, A.; Kore, N.; Vinod, T. K. Unpublished results.









