Reports: UR150675-UR1: Planar Chiral Sulfinyl Diene Iron(0) Tricarbonyl Complexes as a Platform for Diastereoselective Synthesis of Spiroketals
 printer friendly
printer friendlyThe development of the chemistry of enantiomerically pure sulfinyl diene iron(0) tricarbonyl complexes has continued throughout the duration of the grant period. Following our earlier discovery that appropriately substituted complexes of this type can undergo diastereoselective spiroketalizations (Fig. 1), our laboratory has been exploring the substrate scope of this process and as well other related processes.
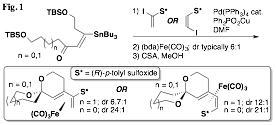
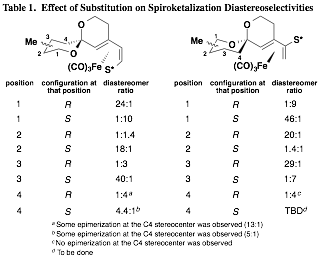
Our initial findings demonstrated that the planar chirality of the substrate was sufficient to influence the formation of the spiroketal stereocenter with a high degree of selectivity. However, it remained unclear whether any additional stereocenters located at positions along either spiroketal ring would play a role in determining the selectivity of the cyclization. While the impact of methyl groups along the two available positions of the A ring was established prior to the grant period, significant effort was next undertaken to complete the study by preparing and inducing the cyclization of substrates with methyl groups along B ring positions. This study is nearly complete at this time, and a clear pattern has emerged. It was found that substrates with "matched" stereocenters (i.e., with methyl groups that would be equatorial) could improve the diastereoselectivity of spiroketalization to as much as 46:1. On the other hand, diastereomeric substrates usually had a preference to invert the spiroketal stereocenter rather than exhibit an axial methyl group. These "mismatched" substrates underwent spirocyclization with little selectivity in some cases, and in others with selectivity as high as 9:1 but dictated by the methyl group's conformational preference to be equatorial rather than by the planar chirality of the iron(0) tricarbonyl complex. The results of this nearly completed study are summarized in Table 1. It should be noted that with the methyl group positioned at C4 of the B ring, avoidance of a non-bonding interaction with the methyl group and the Fe(CO)3 unit was the stereodefining factor. In two of the three cases, epimerization at the C4 stereocenter was observed (the fourth and final case has not yet been completed).
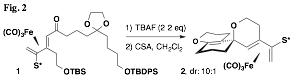
We attempted to extend this concept to the more challenging bisspiroketal analogs but unfortunately have yet been unable to prepare the unsubstituted cases (i.e., without the added stereocenter bearing the methyl group). A major challenge has been to devise a protecting group scheme that is compatible with the key intermediates of the synthetic sequence: the sulfinyl diene, its corresponding complex, and their vinyl stannane precursors. One approach that appeared promising until the final step utilized the keto acetal diol derived from complex 1 (Fig. 2). However, treatment with acid (in anhydrous conditions) instead led to the presumed formation of an intermediate enol ether derived from the acetal; this cyclized on the oxonium ion adjacent to the iron(0) tricarbonyl diene complex. After elimination the unexpected oxaspirocycle 2 was formed; its structure was verified by X-ray crystallography.
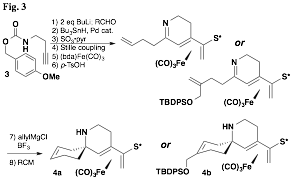
We also initiated a new project aimed at the synthesis of azaspirocyclic systems. The preparation of a cyclic ketimine presented a significant challenge that was met with a strategy that began with a carbamate-protected homopropargylic amine (3, Fig. 3). The dianion generated from it reacted exclusively at the alkyne terminus with an assortment of substituted aldehydes. The ketimines were then prepared in a single step by deprotection of the carbamate and cyclization that followed a sequence of steps in accord with our established methodology. Allylation of this imine proceeded with perfect 100:0 diastereoselectivity, and azaspirocycles 4a and 4b could indeed be prepared in good yield by ring-closing metathesis using the second-generation Grubbs-Hoveyda catalyst. Unfortunately the sequence suffers from the instability of the cyclic ketimines, which decomposes upon standing or even in solution. Future plans include the development of a sequence that would reverse the order that the A and B rings would be prepared.
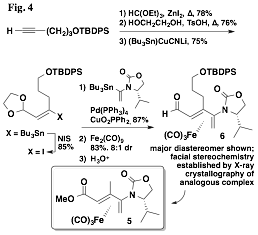
Finally, we have returned to a project, left dormant prior to the grant period, that sought to utilize a chiral auxiliary other than a sulfoxide so that manipulation of the iron tricarbonyl diene complex following cyclizations would more facile and general. We had been exploring the complexation of oxazolidinone dienes, and while the complexations had proceeded with diastereoselectivities similar, or better, than those of the sulfinyl dienes, we had long been unable to ascertain the facial stereochemistry of the major product of these reactions. However, that changed with the synthesis of complex 5 – we were able to obtain X-ray crystal structures of both major and minor diastereomers. With that result we resumed the development of methodologies in this area, and thus far we have developed an approach to oxazolidinoyl dienal iron(0) tricarbonyl complexes exemplified by 6, shown in Fig. 4. We anticipate having the ability to perform diastereoselective additions to the aldehyde that mirror much of the chemistry we developed earlier with the analogous sulfinyl diene complexes.
Five undergraduate students have collaborated with me on these projects; of the three who have graduated, two are enrolled in doctoral programs in synthetic chemistry (at Princeton and at Stanford) and the third is in Japan on a Fulbright Fellowship, working in the Hayashi laboratory at Tohoku University in Sendai. She will be pursuing her doctoral studies at Scripps upon completion of the study abroad. The other two students have both expressed a strong desire to pursue graduate studies in chemistry following their graduation from Swarthmore. As principal investigator, the grant has allowed me to expand the projects available to my students at a key point in my career. The completion of several of these projects is within sight, and will result in publications in the near future; this should enable a successful grant proposal to NSF for the next phase of my career.









