Reports: UNI150667-UNI1: Synthesis and Borate-Catalyzed Kinetic Resolution of Terminal Aziridines
 printer friendly
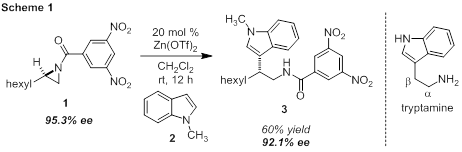
printer friendlyThe ACS Petroleum Research Fund has supported our group's continuing program to advance N-acylaziridines as key intermediates in the synthesis of nitrogen-containing small molecules. In the initial year of funding we observed the stereospecific addition of an indole nucleophile to an enantioenriched N-acylaziridine. Initially, zinc triflate was identified as a competent catalyst for the addition of N-methylindole (2, 4 equiv.) to N-acylaziridine 1 in 60% yield with only a slight loss of enantiopurity (Scheme 1). The 3,5-dinitrobenzoyl (DNB)-protected aziridines performed best in our previous studies on aziridine kinetic resolution. The preference for internal aziridine attack was confirmed by COSY and HMBC studies on tryptamine 3. Beta-substituted tryptamines produced from this route are structurally unique, as alpha-substituted tryptamines are prevalent in nature.
The moderate yield observed under our initial zinc-catalyzed, indole addition reaction conditions is partially linked to a competitive rearrangement process. Therefore, we performed a screen of Lewis acids using an NMR internal standard to determine conversion and product ratios (Table 1, 5 versus 6). N-Acylaziridine 4 was subjected to Lewis acid conditions in the presence of 1H-indole either at room temperature or -78 °C. A series of triflate salts (Table 1, entries 2–9) produced similar ratios of addition to oxazoline as zinc. Our hope was to develop a process employing catalytic amounts of Lewis acid; however, the lack of success identifying an improved metal–triflate salt led us to consider a stoichiometric Lewis acid known to facilitate aziridine opening. Indeed, boron trifluoride (BF3) was the superior Lewis acid promoting ring opening at -78 °C in one hour (entry 10). The reaction was clean with no oxazoline (6) formed, and complete stereochemical transfer to tryptamine 5 was observed. N-Alkylindoles proved more efficient with product ratios favoring the addition product (entries 11–12). Other indoles with more common nitrogen protecting groups were not effective (entries 13–14).
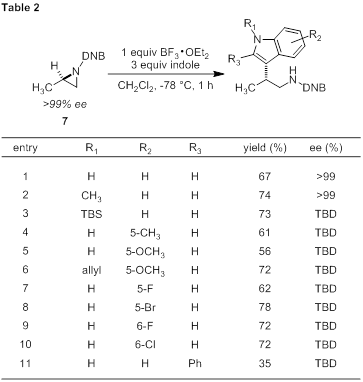
A series of indoles containing diverse functionality was subjected to our BF3-promoted reaction conditions using enantioenriched aziridine 7 (Table 2). The beta-tryptamine products could be isolate in moderate to good yield bright yellow to red solids. We believe the compound colors are related to a charge transfer interaction of the electron-rich indole with the electron-poor DNB group. Free indole and electron rich N-substituted indoles perform well (entries 1–3). The efficiency of N-TBS-indole was encouraging and provides an alternative protecting group strategy to N-alkylindoles. The poor reactivity of N-TBDPS-indole (see Table 1) is likely a steric issue. A variety of electron rich or slightly electron poor substituents are tolerated at the 5- and 6-position of the indole (entries 4–10). While the free indole is preferred as nucleophile, N-allylindoles show an improvement in yield (entry 5 versus 6). Indoles containing substitution at the 2-position show reduced reactivity (entry 11), while substitution at the 4- and 7-position completely shut down reactivity (data not shown). To date, the product ee has only been determined for two products (entries 1 and 2). Based on these results, we believe the reaction will be generally stereospecific; however, more work is needed to confirm the ee for all entries in Table 2.
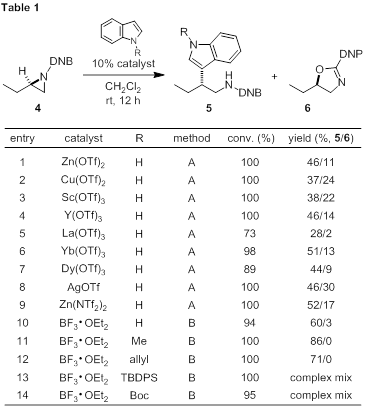
Conditions:
A) 10 mol% catalyst, CH2Cl2 (0.2 M), rt, 24 h
B) 1 equiv BF3•OEt2, CH2Cl2 (0.2 M), -78 °C, 1 h.
In order to investigate the effect of aziridine substitution on reactivity, we required access to a series of 2-substituted N-DNB-aziridines. A library of DNB-protected aziridines was produced in both racemic and enantiopure form to investigate aziridine substrate scope. Ultimately, the deprotection/reprotection of N-tosylaziridines has proven most general for DNB-aziridine synthesis when a succinimide reagent is utilized. N-Tosylaziridines are readily synthesized in both racemic and enantiopure form. Alternatively, N-Cbz-aziridines can be employed in a similar deprotection/reprotection sequence. Enantioenriched N-Cbz-aziridines are available from racemic epoxides in 2 steps or by direct enantioselective aziridination. Terminal DNB-substrates are currently limited to alkyl carbon substituents. Both cis and trans disubstituted aziridines have been synthesized for subsequent ring opening.
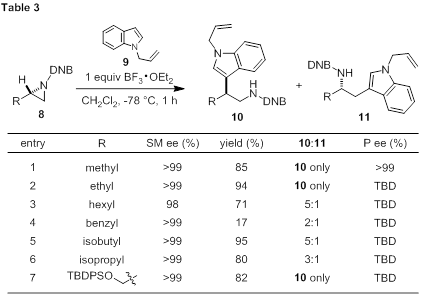
The effect of aziridine structure on nucleophile addition of N-allylindole follows a clear steric trend. N-Acylaziridines (8) containing small R groups undergo highly efficient boron-promoted ring opening in the presence of indole 9 (Table 3, entries 1, 2, 7). Only tryptamine 10 was produced in excellent yield. The reaction for R = methyl is stereospecific (entry 1); however, more work is needed to determine if this trend is general. As the size of the R group increases, the regioselectivity decreases (entries 3–6). The benzyl group is a clear outlier to this trend for reasons that are not yet clear, except numerous side products were produced during the reaction (entry 4). Attempts to employ R = tert-butyl led to little or no formation of tryptamine 10 (data not shown). Moving forward, chiral disubstituted aziridines will be pursued to further test effects of regiocontrol.
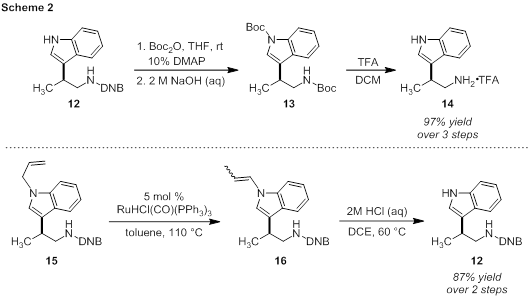
Protecting group manipulation will be critical in processing the b-substituted tryptamine products to more complex bio-active molecules. We have performed initial experiments to this end with tryptamines 12 and 15 (Scheme 2). Complete Boc protection of 12 occurs readily at room temperature. Without workup, the DNB group can be cleaved by treatment with aqueous NaOH. The bis-Boc derivative (13) can be processed without purification to the free tryptamine (14) in excellent yield. By an alternate method, selective indole de-allylation can be accomplished. The alkene in tryptamine 15 is isomerized under ruthenium catalysis to produce an inconsequential mixture of alkene isomers. Subsequent hydrolysis of the vinyl group occurs after heating with aqueous HCl. We are now poised to synthesize more complex tryptamine-based molecules using the methodology developed during the grant period.
In conclusion, support from the Petroleum Research Fund has facilitated the development of N-acylaziridines as linchpin intermediates for the synthesis of enantioenriched beta-substituted tryptamines. During the current grant period, one (1) Master's students and three (3) undergraduate students gained valuable research experience on this project. This data is being used in applications for expanded research funding through the National Institutes of Health and National Science Foundation.









