Reports: DNI352066-DNI3: Cobalt Mediated C-C Bond Transformations toward Small Molecule Synthesis
 printer friendly
printer friendlyTransition metal catalysis which forms or transforms carbon-carbon bonds via oxidative addition and reductive elimination reactions is at the forefront of process development with applications in plastics, pharmaceuticals, materials engineering, and commodity chemical production. Such catalysis is currently dominated by platinum group metals whose relatively high cost, toxicity, and remaining substrate challenges create a need to explore more abundant base-metal surrogates. While catalyst development using a small subset of base-metals (e.g. nickel) has demonstrated incredible potential, development of more attractive metals, such as cobalt and iron, remains underexplored. Our laboratory has recently leveraged the trimethylphosphine-cobalt platform to examine the mechanistic features for key elementary reactions required catalytic C-C bond transformation including C-CN bond oxidative addition, selective two electron C-C bond reductive elimination, and cobalt-to-cobalt exchange of alkyl groups.[1]
Under the past year of funding from the Petroleum Research Fund, our program has completed an near comprehensive mechanistic investigation into the intermolecular exchange of cobalt-methyl ligands in cis,mer-(PMe3)3Co(CH3)2X (X = Cl, I) (1-Me2).[2] The exchange of alkyl groups from one metal to another has received attention due to its relationship to the biosynthesis of acetyl coenzyme A, deleterious scrambling observed during palladium catalyzed cross-coupling, and as a potential step in methane oligomerization techniques. The cobalt-to-cobalt exchange of methyl groups first garnered our interests during experiments to study the pathway for C-C bond elimination at 1-Me2 (Figure 1). Close examination of the reductive elimination reaction revealed isotopic scrambling of the methyl ligands prior to reductive ethane elimination.
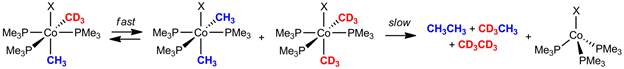
Figure 1.
Mechanistic studies of ethane elimination from 1-Me2 have previously established a rapid and reversible dissociation of the PMe3 ligand trans to Co-Me.1 Given that a related cobalt-to-cobalt alkyl exchange process studied by Bergman and coworkers had implicated ligand dissociation as a mechanistic feature, the role of phosphine loss in the scrambling of 1-Me2 was examined.[3] Surprisingly, the presence of excess PMe3 to the d3-isotopolgoue of 1-Me2 did not substantially alter the rate of 1-Me2 growth in the 1H NMR spectrum. However, control experiments suggest the excess PMe3 opens a second exchange pathway in which the Co-Me and P-Me interchange through a phosphorane intermediate (Figure 2). Such phosphine-metal substituent exchanges are salient to the development of cobalt as a C-C cross-coupling mediator, as similar deleterious exchanges have hampered many heavy metal cross-coupling catalysts.[4]
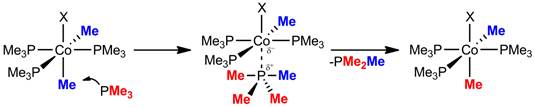
Figure 2.
Given the complexity of two methyl scrambling pathways from 1-Me2, both of which are potentially influenced by the concentration of free PMe3, our laboratory sought other methods to probe the role of ligand dissociation in the cobalt-to-cobalt group transfer reaction. The most productive experiments employed a tridentate chelating model of the tris(trimethylphosphine) complex, (triphos)Co(CH3)2X (2-Me2) (Figure 3). Relative scrambling rate studies between 1-Me2 and 2-Me2 reveal a much slower exchange for the chelating phosphine complex, which indicates the structural barrier opposing phosphine dissociation in 2-Me2 does indeed inhibit intermetallic exchange. Additional crossover studies with 1-Me2 and 2-Me2 are consistent with the required phosphine dissociation occurring at only one cobalt center, leading to a bimolecular methyl transfer event. This data, along with control experiments eliminating radical crossover, reversible methyl halide elimination, and cobalt(I) catalyzed disproportionation pathways have produced a model for the reaction mechanism as illustrated below (Figure 4).
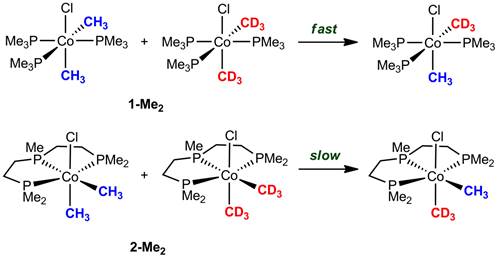
Figure 3.
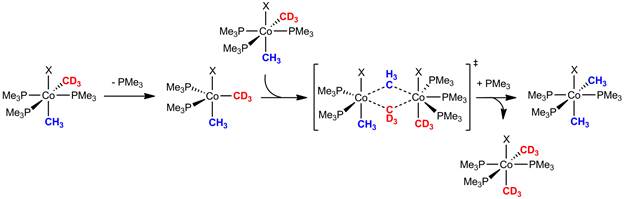
Figure 4.
In summary, this program has elucidated the key mechanistic features of a cobalt-to-cobalt alkyl group transfer reaction. The process occurs through reversible loss of a phosphine ligand, followed by a bimolecular substitution-like transition structure. Over the course of this investigations, a rare, reversible P-CH3 was also identified. Together these finding indicate that, though significant to labeling studies, the alkyl group transfer events are unlikely to meaningfully impact the rates or selectivity of cobalt(III) C-C bond reductive elimination reactions. As part of this funded program, our laboratory is continuing to pursue fundamental insights into other elementary transformations required to utilize cobalt in C-C bond forming and breaking processes.
[1] a) Xu, H.; Bernskoetter, W. H. J. Am. Chem. Soc. 2011, 133, 14956-14959. b) Xu, H.; Williard, P .G.; Bernskoetter, W. H. Organometallics, 2012, 31, 1588-1590. c) Xu, H.; Williard, P .G.; Bernskoetter, W. H. Organometallics, 2013, 32, 798-806.
[2] a) Klein, H.F.; Karsch, H.H. Chem. Ber. 1975, 108, 944-955. b) Klein, H.F.; Karsch, H.H. Chem. Ber. 1975, 108, 956. c) Beck, R.; Klein, H. F. Z. Anorg. Allg. Chem. 2008, 634, 1971.
[3] Bryndza, H. E.; Evitt, E. R.; Bergman, R. B. J. Am. Chem. Soc. 1980, 102, 4948.
[4] Macgregor, S. A. Chem. Soc. Rev. 2007, 36, 67.









