Reports: DNI151975-DNI1: Stereoselective Homoallylation of Aldehydes and Related Compounds
 printer friendly
printer friendlyThe goal of the proposed research is to develop reagents and catalysts for stereoselective homoallylation and homocrotylation (Scheme 1). In the first year of this grant, we have made progress in several areas: 1) asymmetric syn and anti homocrotylation, 2) catalysis, and 3) mechanistic studies.
Scheme 1
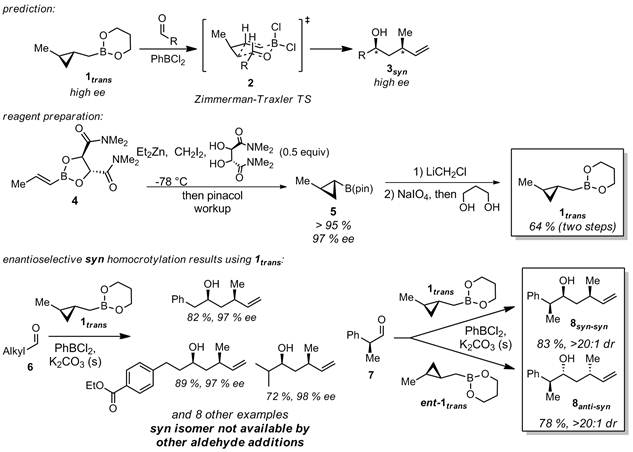
Area 1a) asymmetric syn homocrotylation. Based on our preliminary work,1 our hypothesis was that cyclopropanated crotylboranes homocrotylate aldehydes through Zimmerman-Traxler closed transition states. Thus, we predicted that optically pure 1trans should provide access to optically pure syn products 3. We developed a route (from 4) which conveniently affords gram quantities of optically pure 1trans, and demonstrated a range of syn homocrotylations of aliphatic aldehydes (6) in uniformly high dr and ee. Importantly, no other direct addition to aldehydes can access these products diastereoselectively, let alone enantioselectively. Furthermore, we showed that high selectivity for anti-syn or syn-syn stereotriads (in 8) is obtainable in completely reagent-controlled manner by addition of 1trans or ent-1trans to chiral aldehydes such as 7. These results were the subject of a JACS publication in early 2013.2
Scheme 2
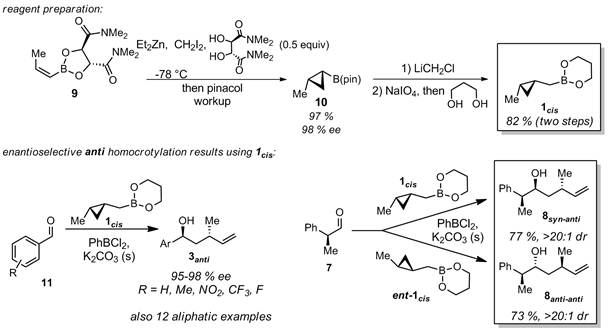
Area 1b) asymmetric anti homocrotylation. We have more recently developed a route to optically pure 1cis, and show that it also adds to aldehydes with uniformly high diastereo- and enantioselectivity (Scheme 2). Whereas additions to chiral aldehydes with 1trans afford anti-syn or syn-syn stereotriads (in 8, Scheme 2), the new 1cis reagent (and ent-1cis) allows us to access anti-anti or syn-anti stereotriads (in 8, Scheme 3), so that all possible stereochemistries of 8 are now accessible from our method. Moreover, we have found that aromatic aldehydes (11) can be homocrotylated in moderate to excellent yields. These results are currently are the subject of a manuscript which is currently in preparation.
Scheme 3
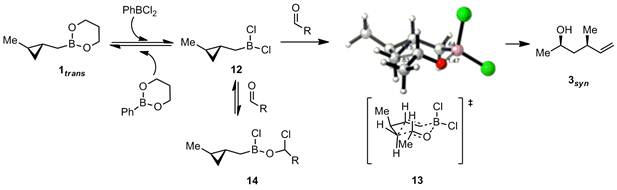
Area 2) mechanistic work. Through NMR studies, we have found that the active homoallylating species present in our reaction mixtures (Scheme 4) is most likely dichloroborane 12, derived from exchange of chlorides and alkoxides between the PhBCl2 activator and the boronate reagent 1. When aldehyde is added to 12, we observe reversible formation of an off-pathway reservoir species 14, which most likely reverts back to 12 before homocrotylation takes place through transition state 13. The proposed transition state 13 is consistent with the observed stereochemical and regiochemical outcomes, and further supported by calculations done in Ken Houk's lab (see ball and stick structure). The tendency to form off-pathway reservoir species 14 likely slows the reaction, and one of the goals of our catalyst design is to eliminate that pathway (see Area 3).
Scheme 4
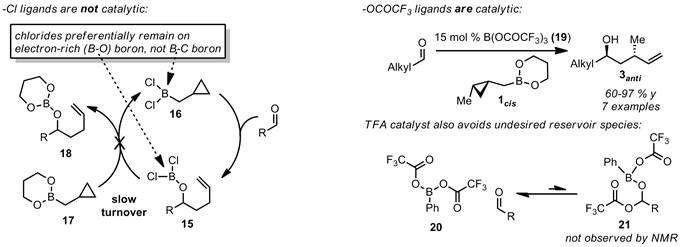
Area 3) catalysis. Based on the above mechanistic work, we speculated that boron carboxylates might be good catalysts for homoallylation. In the PhBCl2-promoted reactions, the unquenched product species 15 is not competent to transfer chlorides to boronate reagent 17 and regenerate 16 to complete the catalytic cycle, likely because chlorides are very electronegative and are most stable on the oxygen-bearing, electron rich boron of 15, rather than on 16. We speculated that, by contrast, trifluoracetates are less electronegative and therefore may be easier to transfer back to the B-C fragment bearing the cyclopropylcarbinyl group (analogous to 16), thus completing the catalytic cycle. Upon testing B(OCOCF3)3 (19), we found that it does operate catalytically, whereas PhBCl2 promotor does not. We can obtain moderate to good yields for a number of homoallylation and homocrotylation products using 15 mol % of this catalyst. Moreover, it seems that the carboxylate-based catalysts do not combine with the aldehydes to form off-pathway reservoir species (21) as does the PhBCl2 promotor (see 14, Scheme 4).
Scheme 5