Reports: UR650687-UR6: Experimental Determination and Bond Energy Decomposition Analysis of Metal-Olefin Bonding Interactions: Towards a Quantitative Metal-Olefin Bonding Model
 printer friendly
printer friendlyDuring the second year, our efforts concentrated on two projects in which computational and experimental progress were made. The first project involved the computation of the metal-olefin bond energies in the series of complexes M(CO)5(C2H4-nXn), with M = Cr, Mo or W; X = CN, and n = 0 to 4, when the cyanoalkeneis bonded via a nitrogen atom in one of the cyano units (h1 binding mode). A full computational study (density functional theory, DFT) of the determination of the bond formation energies for the reaction: M(CO)5 + C2H4-nXn → M(CO)5(h1-C2H4-nXn) have been completed and compared to our previous results in which the alkene was bonded via the double bond (h2 mode).1 The study revealed that the h1- binding mode is thermodynamically preferred. Calculated bond energies for the h1-bonded alkenes did not show strong dependences in the number of cyano substituents and trends agreed well with activation enthalpy trends we measured for the fumaronitrile (trans-dicyanoethyelene) and tetracyanoethylene complexes of the metal complex triad. We attempted to crystallize these complexes but could not obtain crystals suitable for x-ray diffraction analysis. However, the work of Kaim and coworkers2 revealed the preference for the h1-binding mode via an x-ray diffraction report of the W(CO)5(h1-C2(CN)4). Our calculations indicate that the h1-mode is preferred despite the weaker orbital interactions (Figure 1), because the h2- binding mode requires larger steric repulsive interactions and energetically expensive reorganization of the alkene’s geometrical conformation, which is proportional to the amount of the attractive binding orbital interaction.3
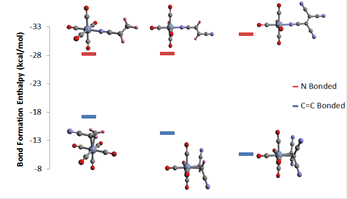
Figure 1. Bond formation enthalpy for N-bonded (h1) and C=C bonded (h2) complexes of Cr(CO)5(cyanoethylene). Shown are complexes with acrylonitrile, fumaronitrile and tetracyanoethyelene.
We have also carried out competitive kinetic determinations of the activation enthalpy of the thermal decomposition of the fumaronitrile and tetracyanoethylene complexes of the chromium and tungsten pentacarbonylcomplexes, although the final data analysis is not complete yet.
The second project involved the continuation of the computational chemistry (DFT) determination of the metal-olefin bond formation energies in the series of complexes M(CO)5(C2H4-nPhn), with M = Cr, Mo or W; Ph = phenyl. This study was intended to show the effects of steric repulsive forces on the metal-olefin bond formation. As expected, an increase in the number of phenyl groups decreases the metal-olefin bond strength. The effect is very dramatic as shown in Figure 2. The calculations predict extremely weak bonds (about 8-13 kcal/mol) for the triphenylethylene complexes and no binding of the tetraphenylethylene complexes.4 Interestingly, one of the calculations derived into a more stable h2- complex formation involving one of the phenyl rings of tetraphenylethylene. We attempted the photo-initiated synthesis of the trans-stilbene complex of chromium pentacarbonyl, but encounter technical difficulties as trans-stilbene absorbs strongly the ultraviolet light from the source. We have started trials in which trans-stilbeneis slowly added to the mixture. Some improvements were made on the yield of what it appears to be the expected product. Improvements on the synthesis are undergoing.
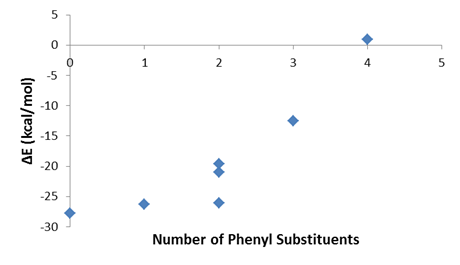
Figure 2. Calculated bond formation energy for W(CO)5(C2H4-nPhn) complexes, Ph = phenyl.
1. Stewart, A.; Johnson, S. L.; Camper, T. W.; Rusk, A.; Cedeno, D. L. “Computational and experimental studies of cyanoethylene pentacarbonyl metal (molybdenum and tungsten) complexes: Insights into η2 and η1 competitive bonding” Abstracts of Papers, 245th ACS National Meeting & Exposition, New Orleans, LA, United States, April 7-11, 2013, CHED-721.
2. Bubrin, M.; Krafft, M. J.; Steudle, L.; Hübner, R.; Field, J. S.; Záliš, S.; Kaim, W. “Structural reassessment of [W(CO)5(TCNE)]: N (σ) coordination instead of an olefin (π) complex” Organometallics 2012, 31, 6305-6311.
3. Cedeño, D. L.; Johnson, S. L.; Stewart, A. B. “Bonding Interactions between a Group 6 Transition Metal Pentacarbonyl and Cyanoethylenes: Why is N-bonding (h1) preferred over olefin (h2) bonding?” Submitted to Organometallicson 08/13/2013. Under peer review.
4. Hon, S. A.; Ryba, K. L.; Cedeno, David L. “Computational study of phenylethylene pentacarbonyl metal (chromium, molybdenum and tungsten) complexes: The influence of steric effects on metal-olefin bonding” Abstracts of Papers, 245th ACS National Meeting & Exposition, New Orleans, LA, United States, April 7-11, 2013, CHED-722.









