Reports: UR652486-UR6: Electron-Induced Reactions: From Methods for Characterization of Intermediates to Mechanisms
 printer friendly
printer friendlyThis research project is about electron-induced reactions, and its two main foci are methods for the characterization of intermediates in these reactions and studying the mechanisms of electron-induced reactions themselves. In the first year three main projects were started:
(1) Systematic comparison of extrapolation methods to estimate the energy – and possibly the lifetime – of electronically metastable states. This idea goes back to the 1960-ties and 1970-ties, but what is missing is, on the one hand, a systematic comparison, with well characterized metastable states, and, on the other hand, a systematic investigations of what can be done with quantum chemistry codes for molecules, say, are potential energy surfaces smooth? This project is currently under investigation in collaboration with Dr. Masahiro Ehara at the Japaneses Institute for Molecular Science. At Southeastern the undergraduate researchers Nicole Blondeau and Robin Joshi worked on it, and preliminary results of the undergraduate research were presented at the 2012 Spring national ACS in the PCHEM poster session.

The Figure shows some of our results for the well known temporary anion of carbon dioxide. Results for its energy converge quickly with the number of diffuse functions in the basis set, and – as expected – much more slowly with the cardinal number of the basis set. However, the error bars estimated from choosing different extrapolation schemes and different weights in the extrapolation procedure are much larger than those expected typically for quantum chemical calculations.
(2) Characterization of dipole-bound and valence anions of para-nitroaniline and larger para-nitropenylalanine systems. This is a collaboration with the experimental groups of Dr. Robert Compton at the University of Tennessee and Dr. Kit Bown at John Hopkins University. These molecules are typical examples that should follow a doorway mechanism, i.e., attachment of an excess electron into a dipole-bound state at thermal energies with rapid transfer into a valence state. Subsequently, the available energy could either be redistributed or the nature of the valence state could prepare cleavage of specific bonds. For the relatively large molecules investigated here, the first step of the PI's, that is, the theoretical contribution, was to calibrate a density functional method for predicting valence states (ab initio methods are either too demanding or unreliable, second-order perturbation theory fails for reason of huge spin contamination). One paper has been published, one is presently under review.
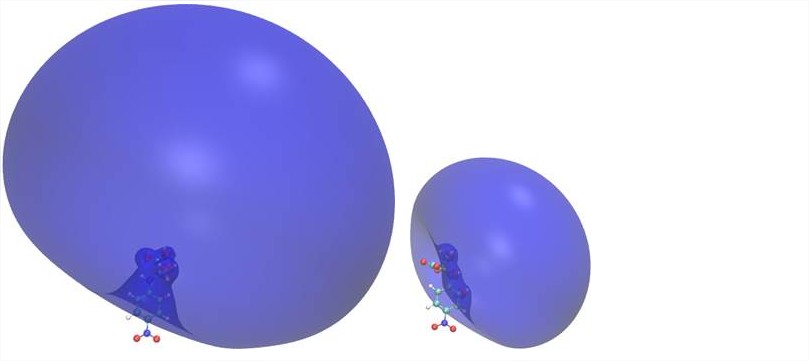
The figure shows dipole-bound states of two conformers of N-para-nitropenylalanine, which have similar dipole moments, yet very different electron binding properties. The reasons are relative orientation of the molecular dipole and volume excluded for the excess electron by the valence density of the neutral.
(3) Development of an analysis scheme for distinguishing the nature of an excess electron regarding its valence or non-valence character. This project was born out of the need to visualize and characterize the distribution of excess electrons in many different species. In contrast to the many existing analysis schemes that partition the total density or that associated with a single orbital into atomic contributions, there does not seem to be any satisfactory method for partitioning it into valance and non-valence density, to assign at least an approximate valence character, or to visualize its properties, say an analog of the atomic radial density, but applicable to molecules without symmetry. It is therefore hardly surprising that there are various controversies in the excess-electron field, which can ultimately be traced back to different measures or questionable interpretations of non-valence character. The first concrete example considered was the excess electron of Li(NH3)4, which was an undergraduate research project of Katelyn Dreux at Southeastern, and was later published in the Journal of Chemical Physics. During the last semester the scheme was generalized, and put on solid mathematical footing, and a manuscript has been submitted to the Journal of Chemical Theory and Computation.
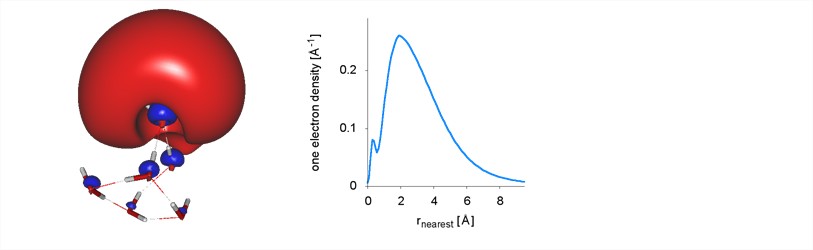
The figure shows an iso-surface representation (left) of a “surface-bound” electron of a water cluster and the associated distribution function (right), which allows one to assign a valence character of less than 10% to it.
The impact of this research and this grant on my career and on that of my student can hardly be overstated. Needless to say that at a primary undergraduate institution any meaningful professional development without time and funds for summer research is a challenge. But especially in these times of ever tighter budgets even activities once taken for granted, such as making presentations at regional meetings, become increasingly difficult. Thus, this grant is a key stone for maintaining an active and professional research program as such.
For my students the same is true. To them it makes a huge difference to know that they do not just participate in some educational project, but in a real research project that may at some point land them a poster presentation or even a co-authorship on a paper. They take their work more seriously, begin to see the value of a deeper level of understanding, and are more likely to continue “their” research after the mandatory classes have been taken. Katelyn has in fact entered the graduate program at the University of Mississippi, where she is working towards a PhD in Theoretical Physical Chemistry.









