Reports: UNI351903-UNI3: Biomimetic Catalysts for Hydrogen Production and Carbon Dioxide Activation
 printer friendly
printer friendlyIntroduction
The proposed research focuses on Cu-Mo binuclear complexes and similar organometallic compounds that mimic the Cu-Mo metallocluster found in the active site of carbon monoxide dehydrogenase (CODH) from O. carboxidovorans. The role of the Mo-Cu metal cluster found in the active site of CODH is to catalyze the reversible oxidation/reduction of CO to CO2 (eq.1).
![]()
While the forward reaction is relevant for hydrogen (H2) production and similar to water gas shift (WGS) reaction, the reverse reaction could be applied to convert carbon dioxide (CO2) into useful products such as methanol and dimethyl ether. Our proposal had two primary goals: (1) to investigate the structural and mechanistic characteristics of the Mo-Cu binuclear catalysts for H2 production and rationally design analogue catalysts for both H2 production and CO2 activation by employing molecular modeling and electronic structure calculations; (2) to assess the charge distribution within the metal cluster and the redox-active nature of various ligands that could influence the reactivity of metal centers. The work carried out in the first year of the PRF grant has focused on both goals.
Results
The proposed methodology (goal 1) consisted of a series of calculations that generated energy profiles of the catalytic mechanism for different metal clusters. While the active site of CODH contains a Mo-Cu cluster, we have also tested W-Cu, Mo-Ag, and W-Ag metal clusters while preserving the non-innocent dithiolene ligand, which is a smaller model of the molybdopterin cofactor present in the native structure of CODH, and replacing it with another non-innocent ligand, diimine. During this past year, we have completed all of these eight energy profiles using density functional theory and pseudo-potential basis sets (PBE0 with 6-311++G(d,p) for H, C, N, and O; modified LANL2DZ for all transition metals; LANL2DZ augmented with diffuse functions for S). We have also tested different environments for the catalytic sites, such as amino acids that resemble enzyme surroundings and explicit water molecules that stabilize the open conformation of this large organometallic complex. The most promising results were obtained for W-Cu metallocluster with a dithiolene ligand (Figure 1). This catalyst formed more reactive intermediates that effected in lower energy barriers. Also, the thermodynamics are more favorable for both forward and reverse WGS reactions. The problem with the original Mo-Cu cluster was the formation of a very stable intermediate during the CO2 formation (-44 kcal/mol for Mo-Cu as compared to -15 kcal/mol for W-Cu). A similar stable intermediate was also formed with the Mo-Ag catalyst (-42 kcal/mol). The W-Ag catalyst showed a moderately stable intermediate (-21 kcal/mol).
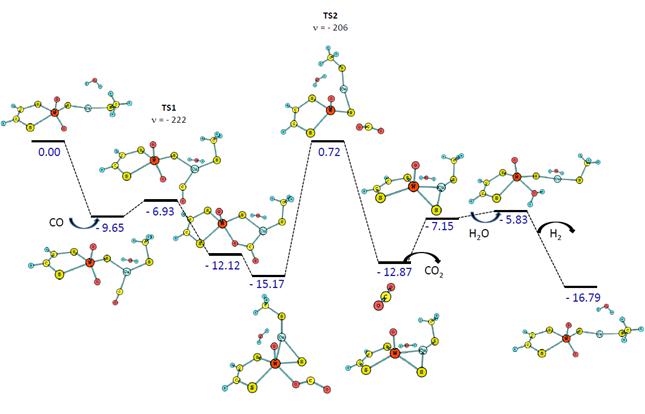
Figure 1. Energetics (enthalpy in kcal/mol) of the WGS reaction pathway using a catalyst that contains W-Cu metallocluster and dithiolene ligand.
The presence of the diimine ligand in analogue catalysts resulted in less stable intermediates during the CO2 formation (-15 to -7 kcal/mol for all four metal-metal combinations), but the thermodynamics were unfavorable during the elimination of CO2 and/or binding of H2O. We are currently studying the influence of non-innocent oxalate ligands on the energy profiles of WGS reaction mechanisms.
The work carried out in the first year of the PRF grant has also focused on understanding the electronic distribution in the catalytic core of the proposed organometallic complexes (goal 2). We employed Natural Bond Order (NBO) calculations that revealed that the bridging S atom present in the metallocluster has a role in the electronic communication between the two metal centers. When dithiolene ligand is bonded to the Mo metal center, there is a strong covalent interaction between Mo and all three S atoms (bridging S and dithiolene S atoms) in the singlet state. The axial oxygen atom is weakly bound (mostly electrostatic interaction with a small covalent component). When triplet state was analyzed, the axial O atom was more strongly bonded to Mo while the bridging S atom was more weakly bonded to Mo. A similar electron configuration was observed after CO bonded to the Cu center. These behaviors suggest that singlet states will be dominant. Electronic structure calculations confirmed that singlet states are more favorable. If CO is inserted between Cu and bridging S (relevant to the very stable intermediate during CO2 formation), the Mo-S covalent interactions become weaker, allowing the formation of the stable thiocarbamate complex. When Mo is replaced by W, the soft-soft interactions between W and S atoms are stronger, resulting in a more favorable reaction pathway as illustrated in Figure 1. A similar effect was observed with diimine ligands; the N-Mo bonds were weaker, while Mo-O and Mo-S become stronger. Because of these electronic configurations, the thiocarbamate complex was more weakly bonded, resulting in a less stable intermediate.
Conclusion
While the results from the first year of the PRF grant are encouraging, we hope to further develop these projects with an emphasis on studying a larger range of catalysts displaying predictable activities.
Student Participation and Dissemination
The manuscript that will communicate these results to the chemistry community is in preparation. There will be six undergraduate student authors from Wilkes University. Funds from the ACS-PRF have been used to support five of them during the summers of 2012 and 2013. Of the student authors that have been supported by ACS-PRF, two will graduate in 2014 and their intentions are to pursue careers in secondary education and chemical industry, respectively. These students have given poster presentations at national ACS meetings, and oral presentations are scheduled for this fall at a research symposium organized by King's College, Misericordia and Wilkes Universities. The other three undergraduate students are in their second or third year at Wilkes University.









