Reports: UNI350980-UNI3: Ambiphilic Scaffolds for Cooperative Metal-Ligand Activation of Small Molecules
 printer friendly
printer friendlyThe first goal of this project was to develop reliable syntheses for ligand precursors that could be incorporated into such a complex. We chose [RP2Si]H2 ligands with phenylene backbones (e.g., complex 1 in Scheme 1, where R = phenyl or cyclohexyl) as a set of initial targets due to the ready accessibility of o-brominated phenyphosphines through established cross-coupling methods and previous reports indicating that such complexes are useful precursors for multidentate phosphinosilyl ligands. We have developed two successful routes to the synthesis of the diaryl dihydrosilanes: (1) substitution at Si(OEt)4 followed by reduction with lithium aluminum hydride (or deuteride), and (2) substitution at the H2SiCl2 surrogate, (teeda)SiH2Cl2. Both routes have been effective for synthesis of the ligand precursor, and the first route has the advantage of allowing facile synthesis of the dideuterated version of the ligand.
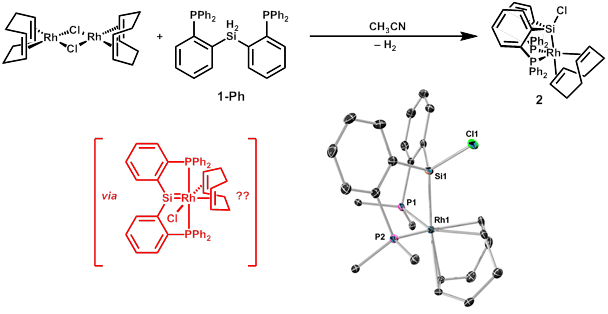
Preliminary studies of the coordination chemistry of 1 have shown that it will form complexes with nickel, cobalt, rhodium, and iridium, though the iridium reactions frequently give numerous products, occasionally involving hydride transfer from silicon without ligation. From the reaction of 1-Ph with [Rh(cod)Cl]2 in acetonitrile, we have identified the 1,5-cyclooctadiene adduct 2 in Scheme 1 as a major product resulting from dehydrogenation and chloride migration to Si. Though the mechanism by which 2 forms is not yet clear, its formation supports our hypothesis about silicon serving a non-innocent coordination role and may implicate a silylene intermediate. We are hopeful that reactions of 1 with cationic rhodium precursors lacking halides will allow access to rhodium silylene pincer complexes by a similar dehydrogenation reaction, and we are also exploring the use of Rh(I) alkyl complexes as potential silylene precursors.
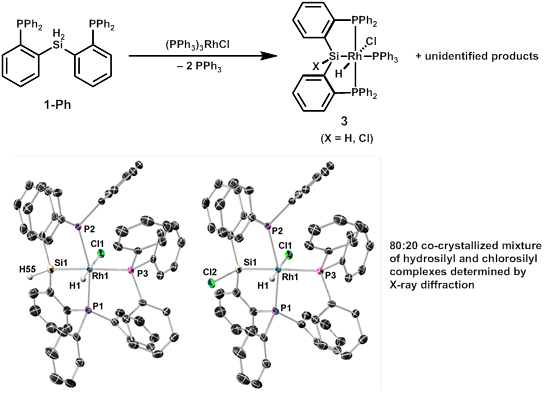
We speculated that we might be able to intercept an intermediate species prior to H2 loss if the reaction in Scheme 1 were performed with a phosphine- rather than diene-supported rhodium center. In fact, use of Wilkinson's catalyst as a starting materials does seem to prevent H2 loss, but we have still seen the formation of chlorosilyl ligands, as evidenced by NMR and an X-ray structure obtained that shows an 80:20 disordered mixture of hydrosilyl and chlorosilyl products (Figure 1). This finding also strongly suggests the non-innocence of the silyl/silylene ligands, and we are exploring what conditions can be used to favor the formation of either hydrosilyl or halosilyl products.
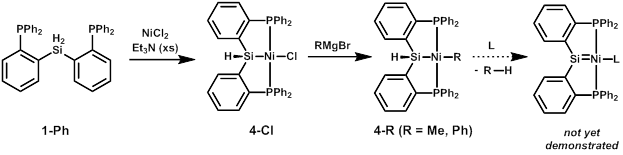
Another route that might minimize competitive halide migration involves using nickel. In this case, metalation in the presence of sacrificial base (Et3N) appears to lead to elimination of HCl and formation of the diamagnetic four-coordinate complex 4-Cl (Scheme 2). Reactivity studies have recently begun, and we have found that nickel complex 4-Cl reacts cleanly with Grignard reagents MeMgBr and PhMgBr to give Ni(II) alkyl complexes 4-Me and 4-Ph. We hope to use these as precursors to nickel silylene complexes via elimination of methane or benzene, respectively, though this route has not yet proven successful.
Upon obtaining the desired metal silylene complexes, reactivity with both nonpolar (e.g., C–H) and polar (e.g., C=O) multiple bonds will be explored with the goal of developing catalysis using the silylene (or hydrosilyl) as a hydride shuttle in reductive or oxidative catalysis with a particular focus on petroleum feedstocks.
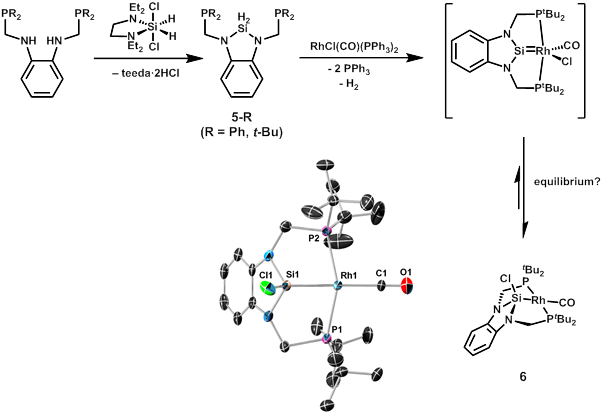
In addition to the results presented above, we have recently discovered a successful preparation of pincer-type N-heterocyclic analogues of the aryl-substituted ligands discussed above. Although N-heterocyclic silylenes (NHSis) have been known for some time, oversubstitution at metal complexes is a frequent problem, hindering their application as spectator ligands or in cooperative catalysis. We have proposed that NHSi complexes may be prepared and stabilized by incorporation of the NHSi unit into a pincer-type ligand like those described above. Accordingly, we adopted the synthetic route outlined in Figure 2, which affords the diamino dihydrosilyl ligand 5-R in good yields (R = Ph, t-Bu). This ligand preparation again makes use of the (teeda)SiH2Cl2 reagent, and it works particularly well since the coordinated teeda serves as an internal base to remove the two equivalents of HCl generated during the reaction.
We have just begun exploring the coordination chemistry of ligands such as 5-R, and they generally react more cleanly than diaryl dihydrosilyl ligands 1-Ph and 1-Cy. In an exciting result that is related to our findings presented above, we have noted that reaction of 5-tBu with RhCl(CO)(PPh3)2 cleanly affords diaminochlorosilyl complex 6, again by dehydrogenation through a putative rhodium silylene intermediate. Complex 6 has also been characterized by X-ray crystallography and we are exploring routes to forming cationic and neutral rhodium silylenes from 5 as well as the potential of NHSi pincer complexes derived from 5 to react in cooperative fashion with small-molecule substrates.