Reports: DNI351009-DNI3: Palladium(I) and Nickel(I) Bridging Allyl Dimers for the Catalytic Functionalization of CO2
 printer friendly
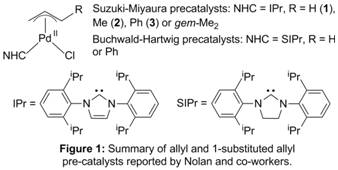
printer friendlyPd-catalyzed cross-coupling is an area of intense research interest because it has numerous applications in the synthesis of pharmaceuticals, fine chemicals, and materials. A major advance in this area has been the use of well-defined, preligated Pd(II) pre-catalysts which can readily generate the monoligated active Pd(0) catalysts. Nolan and co-workers have developed several classes of Pd pre-catalysts supported by bulky NHC ligands, including complexes of the type Pd(NHC)(η3-allyl)Cl (Figure 1). In 2006, they reported that the addition of substituents in the 1-position of the allyl ligand resulted in a dramatic improvement in efficiency. At this stage there is no clear explanation for why cinnamyl substituted species are better pre-catalysts.
Recently, several studies have reported Pd(I) μ-allyl dimers supported by sterically bulky phosphine ligands are viable pre-catalysts for various cross-coupling reactions. However, despite the close relationship between complexes of the type (L)Pd(h3-allyl)Cl and their Pd(I) μ-allyl dimer analogues, little has been done to compare their efficiencies as pre-catalysts. Moreover, the activation of Pd(I) μ-allyl dimer pre-catalysts to a monoligated L-Pd(0) species has not been studied. Given our interest in the chemistry of Pd(I) μ-allyl dimers we have performed a detailed study comparing the reactivity of the Pd(II) pre-catalysts with their corresponding Pd(I) μ-allyl dimers.
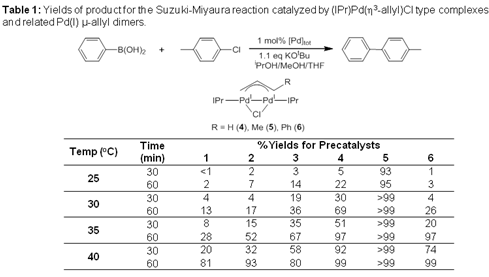
In order to probe the role of dimers in cross-coupling, we synthesized the IPr supported Pd(I) μ-allyl dimer congeners to Nolan's monomeric system through the reaction of 1-3 with a slight excess of K2CO3 in warm ethanol. This new synthetic route is a vast improvement on our previous preparation of Pd(I) m-allyl dimers, which required the formation of air and moisture sensitive intermediates. The catalytic efficiency for the Suzuki-Miyaura reaction of the Pd(II) monomers and their Pd(I) μ-allyl dimer congeners was compared (Table 1). At lower temperatures, there is a large discrepancy between the activity of monomeric 3 and its dimeric congener 6. This implies that under the reaction conditions 3 is not converted into 6 and that activation of 6 is more difficult than activation of 3. The unsubstituted allyl species (1 and 4) were unreactive at 25ºC, but both became more active for catalysis at elevated temperatures, with the dimer always outperforming the monomer. This is consistent with the monomer being converted to the dimer under the catalytic conditions. Under our reaction conditions the cinnamyl monomer 3 greatly outperforms the crotyl monomer 2. In contrast the crotyl dimer 5 is a significantly better pre-catalyst than the cinnamyl dimer 6, and also the unsubstituted allyl dimer 4. The observation that the substitution on the allyl ligand has an effect on the catalytic activity of the dimers prompted us to probe the activation of Pd(I) m-allyl dimer pre-catalysts.
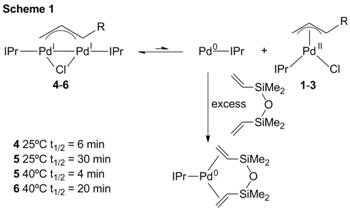
We propose that 4-6 are activated through a disproportionation reaction, in which Pd(IPr)(η3-allyl)Cl (1-3) and catalytically active IPr-Pd(0) are formed. The disproportionation is thermodynamically unfavorable but can be driven by trapping the Pd(0) product. We studied the rates of disproportionation of 4-6 by trapping the Pd(0) with excess 1,3-divinyl-1,1,3,3-tetramethyldisiloxane (dvds) to generate Pd(IPr)(dvds) (Scheme 1). The rate of trapping is 4 > 5 > 6. This suggests that increasing the steric bulk in the 1-position of the allyl ligand increases the transition state energy for disproportionation, although an alternative explanation is that the 1-substituted dimers are thermodynamically stabilized relative to the unsubstituted dimer.
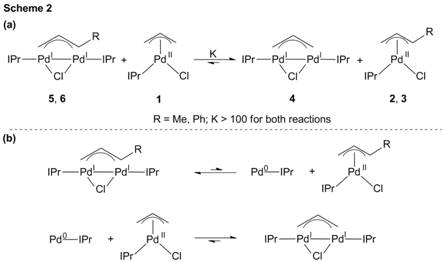
In catalysis using dimers, the rate of disproportionation to form the active IPr-Pd(0) species is important for high activity. However, in monomeric systems dimer formation from comproportionation of IPr-Pd(0) and the starting precatalyst will reduce activity. To probe the relative kinetic barriers for comproportionation to form the dimers, we conducted crossover reactions between unsubstituted 1 and the substituted dimers 5 and 6 (Scheme 2a). The crossover reactions are the combination of the disproportionation of the substituted dimers and the comproportionation of 1 with IPr-Pd(0), as shown in Scheme 3b. The equilibrium strongly favors the formation of 4 from the substituted allyl dimers 5 and 6. This indicates that the comproportionation reaction to form 4 is more exergonic than the comproportionation reactions to form 5 or 6, which implies that the increased barrier to disproportionation in 5 and 6 compared to 4 (Scheme 2) is not due to thermodynamic stabilization of the dimeric products but to an increase in the energy of the transition state. Given that comproportionation is the microscopic reverse of disproportionation, we can thus conclude that the relative barriers for comproportionation in the 1-substituted systems must also be higher than that for the unsubstituted system. This suggests that when the 1-substituted monomers such as 3 are activated, oxidative addition can outcompete comproportionation, thus maximizing the concentration of IPr-Pd(0) that enters the catalytic cycle. At higher temperatures the Pd(I) μ-allyl dimer pre-catalysts are activated via disproportionation to a (IPr)Pd(h3-allyl)Cl species and the IPr-Pd(0) active catalyst, and the monomers and dimers exhibit similar catalytic activity.
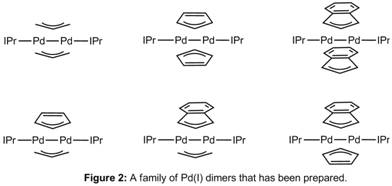
It has previously been postulated that Pd(I) dimers with bridging cyclopentadienyl (Cp) or indenyl ligands are analogous to those with bridging allyl ligands, however the reactivity of these species has not been studied in detail. Last year we synthesized a family of compounds containing different combinations of bridging allyl, Cp or indenyl ligands with ancillary PEt3 ligands. This year we have synthesized and crystallographically characterized the same family of complexes supported by the NHC ligand, IPr (Figure 2). Surprisingly, there are many differences between the IPr supported compounds and the PEt3 compounds. For example, the IPr supported compounds are thermally stable, whereas the PEt3 compounds decompose in both solution and the solid state at room temperature. As a result, in the future we should be able to easily study the reactivity of the IPr supported complexes.