Reports: DNI152183-DNI1: Sustainable Catalysts for Feedstock Chemical Functionalization
 printer friendly
printer friendlyHerein are reported the advances we have made with support from the ACS Petroleum Research Fund during the last year in two areas: (i) urea-activation of diazo compounds and (ii) chiral silanediols in anion-binding catalysis.
Urea Activation of Diazo Compounds. Pioneering discoveries from our laboratory have revealed that ureas are able to elicit and control carbene-like reactivity of diazo compounds. We were initially inspired by the power of the N–H insertion methodology of metal carbenoids1 (eq 1) and set out to investigate an organocatalytic approach for the insertion of diazo compounds into N–H bonds of anilines (eq 2, Scheme 1). Previous work demonstrating the compatibility of diazo compounds with Brønsted acid catalysts added support to our pursuit.2 Our hypothesis was based on exploitation of the urea-nitro group recognition to facilitate the loss of nitrogen gas from 1 to generate a reactive intermediate capable of producing useful products (3 and/or 4) as the result of an N–H insertion reaction or multicomponent coupling. Indeed, the urea-catalyzed N–H insertion/arylation of nitrodiazoesters was effective and multi-component coupling readily occurred to afford the corresponding a-aryl glycine products.3 In our laboratory, we have constructed over 40 a-aryl glycines, incorporating a variety of anilines and indoles, in high yields with this method.
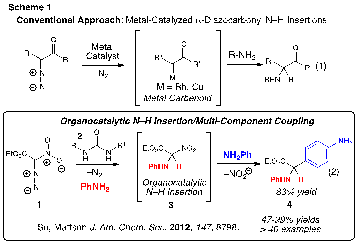
The proposed reaction pathway for formation of a-aryl glycine 4 begins with boronate urea-activation4-6 of the nitrodiazoester through formation of hydrogen-bonded complex I (Scheme 2). By 1H NMR spectroscopy we have learned the binding constant (Ka) for I is 249 M-1 in acetone-d6, a value that suggests the nitronate ion character of 1 is an important factor in binding. The reaction of I with aniline affords N–H insertion adduct II. Our investigations suggest it is likely that the conversion of I to II occurs through a polar transition state instead of a carbene-like transition state. The loss of nitrous acid from II and reaction with a second nucleophile, such as aniline, is proposed to yield the isolated aryl glycine products. In depth studies of the details of the mechanism, including both experimental and computational work, were conducted with assistance from our collaborators in Professor Christopher Hadad's and Professor Jovica Badjic's research groups. This work has recently been submitted to the Journal of the American Chemical Society for publication.

The discovery of our N–H insertion/arylation of nitrodiazoesters prompted us to further explore urea-catalyzed multicomponent couplings of diazo compounds. During our investigations we found nitrodiazoesters (1) undergo unsymmetrical double arylation reactions under the influence of a unique urea and aniline cocatalyst system (Scheme 3). This new reaction enabled the preparation of a large family of geminal diaryl esters (5), valuable compounds in complex target synthesis, in high yields. The optimized reaction conditions found boronate urea 2a and 4-fluoroaniline to be the ideal pair for cocatalysis. For example, 5a was prepared in 86% yield from the coupling of ethyl nitrodiazoacetate, 5-bromoindole and 5-methoxyindole under the influence of urea and aniline cocatalysis. Importantly, no desired diarylated products are prepared when traditional transition metals, like rhodium and copper, are used as catalysts: this demonstrates that urea catalysis enables unique reactivity patterns of diazo compounds. Our current objectives include developing enantioselective urea-catalyzed insertion reactions of diazo compounds and exploring the mechanism of urea-catalyzed double arylation reactions of nitrodiazoesters.

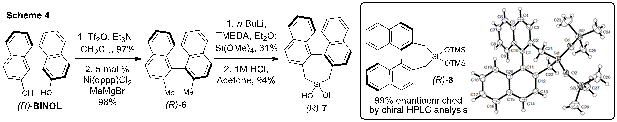
Chiral Silanediols in Anion-Binding Catalysis. Inspired by our success with achiral silanediol hydrogen bond donor catalysis,7 we turned our attention toward the design and syntheses of chiral silanediol catalysts. Our first enantiopure silanediol was prepared from (R)-BINOL (Scheme 4). The triflation of (R)-BINOL8 followed by a Kumada cross-coupling reaction9 gave rise to (R)-2,2'-dimethyl-1,1'-binaphthalene 6 in high yield. Dilithiation of (R)-6 followed by treatment with tetramethoxysilane afforded an intermediate dimethoxysilacycle that readily converted to silanediol (R)-7 upon treatment with hydrochloric acid in acetone. An X-ray quality crystal of bis-trimethylsilyl protected (R)-7 was obtained from hexanes and the ORTEP representation is depicted in Scheme 4.
The exploration of silanediol anion-binding catalysis was initiated with the activation of in situ generated N-acylisoquinolines for reaction with silyl ketene acetals, a reaction with known benefits in the presence of (thio)urea catalysis (Scheme 5).10 It is proposed the silanediol catalyst activates 8 for reaction via ion-pair 9. We were delighted to find that the use of enantiopure silanediol catalyst (R)-7 enabled us to control the absolute stereochemistry of N-acyl Mannich reactions (Scheme 5). For example, in the presence of 20 mol % of (R)-7 product 10 (where X=NO2) was isolated in 72% yield as a 75:25 mixture of enantiomers. A plausible transition state (11) benefits from hydrogen bonding and p-p stacking. Investigations are currently underway to better understand the mechanism of silanediol ion-pair catalysis, including a correlation of silanediol structure to activity, to enable better control over these processes.

Select References.
(1) Moody, C. J. Angew. Chem. Int. Ed. 2007, 46, 9148.
(2) Johnston, J. N.; Muchalski, H.; Troyer, T. L. Angew. Chem. Int. Ed. 2010, 49, 2290.
(3) So, S. S.; Mattson, A. E. J. Am. Chem. Soc. 2012, 134, 8798.
(4) So, S. S.; Auvil, T. J.; Garza, V. J.; Mattson, A. E. Org. Lett. 2012, 14, 444.
(5) So, S. S.; Burkett, J. A.; Mattson, A. E. Org. Lett. 2011, 13, 716.
(7) Schafer, A. G.; Wieting, J. M.; Mattson, A. E. Org. Lett. 2011, 13, 5228.
(8) Page, P. C. B.; Buckley, B. R.; Blacker, A. J. Org. Lett. 2004, 6, 1543.
(9) Kasak, P.; Putala, M. Tetrahedron Lett. 2004, 45, 5279.
(10) Taylor, M. S.; Tokunaga, N.; Jacobsen, E. N. Angew. Chem. Int. Ed. 2005, 44, 6700.









