Reports: UNI152391-UNI1: Stereoselective N-Heterocyclic Carbene (NHC) Catalyzed Oxidopyrylium-Enolate [5+2] Cycloadditions towards Bridged, Polycyclic Ethers
 printer friendly
printer friendlyCycloadditions constitute an efficient avenue toward the synthesis of important ring structures. Whereas Diels-Alder [4+2] cycloadditions are among the most ubiquitous of all organic reactions, [5+2] cycloadditions are significantly less recognizable. However, polycyclic ethers are among the most common classes of ring systems isolated from the natural world. Oxidopyrylium-alkene [5+2] cycloadditions provide structures of high utility, namely bridged polycyclic ethers, which are common to many biologically active natural products such as englerin A, platensimycin, and the cortistatins.

Many strategies toward oxidopyrylium intermediates have been disclosed, but thermal or base-mediated conversion of acetoxypyranones 2 derived from Achmatowicz oxidative rearrangement of readily available furfuryl alcohols 1 endures as a practical and versatile pathway. Intramolecular variants were first reported by Sammes to deliver bridged tricyclic ethers 4 via oxidopyrylium intermediates 3. Jacobsen recently disclosed an elegant approach toward enantioselective, intramolecular [5+2] cycloadditions of similar substrates utilizing hydrogen-bond-donor dual catalysis between achiral and chiral thioureas. Although several examples of acetoxypyranone-based [5+2] cycloadditions toward natural products have been reported, the need for innovations toward these important heterocycles persists.
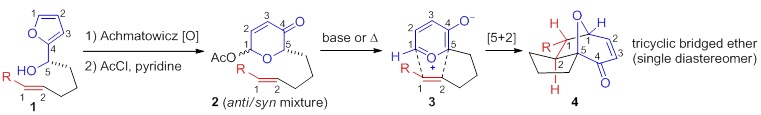
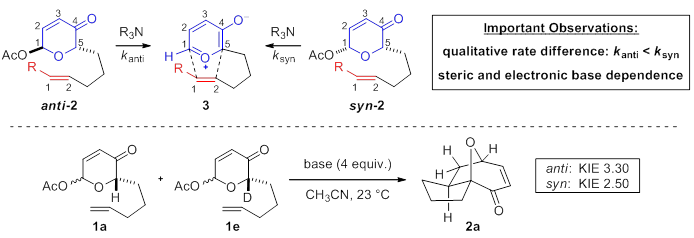
We have discovered unique reactivity of anti- and syn-acetoxypyranones in oxidopyrylium-alkene [5+2] cycloadditions leading to a novel cascade sequence affording a caged tetracyclic lactol (vide infra). The subtle interplay between acetoxypyranone conformation and steric bulk of the tertiary amine bases cause syn-acetoxypyranone 2 to undergo [5+2] cycloaddition appreciably faster than anti-acetoxypyranone 2. We proposed that deprotonation was involved in the rate-determing step and thus studies were undertaken that revealed primary Kinetic Isotope Effects for both 2H-labeled acetoxypyranones anti-1e and syn-1e. This provided evidence that abstraction of H(D) is involved in the rate-determining step suggestive of early transition states that resemble low energy conformations of anti-1a and syn-1a, respectively.
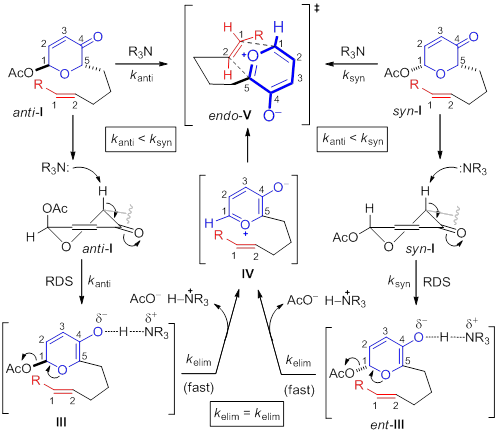
Based on this data, a proposed reaction pathway can be envisioned. Both anti- and syn-I presumably adopt conformations in which the α-proton is coplanar with the π-system of the ketone. Rate-determining deprotonation delivers dienolates III and ent-III that likely have very short lifetimes due to the impending aromaticity. The enantiomeric relationship between these dienolates III and ent-III gives further credence to the proposed rate-determining deprotonation since they would rapidly coalesce thus essentially destroying the inherent difference and bias between acetoxypyranone diastereomers anti- and syn-I. However, it is also important to consider the possibility of slightly different reaction pathways. For example, it is conceivable that formation of oxidopyrylium IV via anti-I, although slower, is more concerted due to the anomeric orientation of the acetate moiety. In either case, elimination of the acetoxy would deliver the oxidopyrylium IV, which undergoes endo [5+2] cycloaddition (i.e. endo-V).
Additionally, the efficiency of a novel cascade process that afforded caged tetracyclic lactol 5a from acetoxypyranone-enal 2a (obtained via cross-metathesis with crotonaldehyde utilizing Grubbs-Hoveyda 2nd Generation catalyst) was determined to be dependent on the relative stereochemistry of each diastereomer (i.e. anti vs. syn), the amine base utilized, and the addition of water.

Unique reactivity of anti- and syn-acetoxypyranone diastereomers was demonstrated in oxidopyrylium-alkene [5+2] cycloadditions. Due to subtle interplay between acetoxypyranone conformations and steric bulk of tertiary amine bases, syn-acetoxypyranones consistently undergo cycloaddition faster than the corresponding anti-acetoxypyranones. Additionally, the efficiency of an oxidopyrylium-alkene [5+2] cycloaddition conjugate addition cascade process that afforded a novel tetracyclic lactol was determined to be dependent upon the relative stereochemistry of the acetoxypyranone, the amine base utilized, and the addition of water. Further studies directed at deeper understanding of this process and synthetic applications are currently underway in our laboratory.
This award has significantly impacted our research in several ways. First, the obvious benefit of funding is that it is a lifeline for research, whether to purchase chemicals or pay students. Indeed, several students (B.S. and M.S.) have benefited from this funding and were given the opportunity to conduct full-time research during the summer months. Finally, the flexibility to pursue serendipitous discoveries that differ from initial proposals is greatly appreciated and has changed the course of our research program for the better.









