AmericanChemicalSociety.com
Reports: AC1 47591-AC1: Asymmetric Hydroamination Catalyzed by Non-Metallocene Complexes of the Group III and Group IV Metals
The catalytic hydroamination of alkenes constitutes a synthetic route to amines that is endowed with exceptional atom economy. In contrast to catalysis by complexes of the late transition metals, hydroaminations involving complexes of the group 3 metals proceed via syn-addition of the metal-amido (M-N) bond to the C=C double bond. In addition, diastereoselectivities associated with intramolecular hydroaminations of chiral substrates by group 3 metals are correlated with both the covalent radius of the metal and its ligand environment. We have previously shown that a non-metallocene complex of Sc(III) supported by a strongly electron donating chelating diamide ligand (e.g., 3) provides superb trans/cis selectivities in the cyclization of 2-substituted-4-pentenamines. During the preceding funding period, we developed an efficient synthesis of (±)-xenovenine (1) that relies on rate enhancement attributable to an unprecedented Sc(III) catalyzed internal hydroamination involving a 2-(5-ethyl-2-thienyl)ethenyl terminator, that serves as a surrogate for a conventional alkyl group.
The toxic pyrrolizidine alkaloid (+)-xenovenine (1) was isolated from the skin of the frog Solenopsis xenoveneum in 1980. From a biological standpoint it is noteworthy that this defense alkaloid is not produced by the frog but instead originates in the venom of ants that constitute, in part, the diet of the amphibian.
We have previously reported that 5-amino-1,8-nonadiene (2a) undergoes smooth sequential bicyclization to pyrrolizidine 5a via pyrrolidine 4a in the presence of complex 3 (5 mol%). The most direct and obvious route to (±)-xenovenine (1) should therefore entail an analogous bicyclization of aminodiene 2b. Unfortunately, common 1,2-disubstituted alkenes are known to be very reluctant participants in internal hydroaminations catalyzed by group 3 complexes. This indeed proved to be the case as 2b, although subject to facile monocyclization to provide 4b (t/c = 49:1, characterized as the p-toluenesulfonamide),
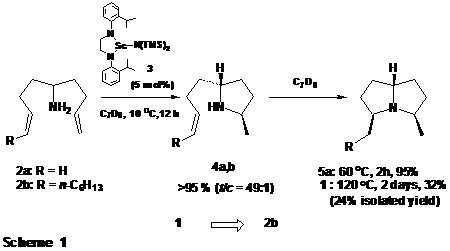 |
underwent the required subsequent cyclization only with great lethargy and incompletely at 120 oC (Scheme 1).
We have previously shown that a representative primary amine bearing a 2-(phenyl)ethenyl moiety (e.g., 1-amino-2,2-dimethyl-5-phenyl-(E)-4-pentene) undergoes internal hydroamination at a significantly faster rate than simple 1,2-disubstituted aminoalkenes (i.e., 1-amino-2,2-dimethyl-(E)-4-hexene). In light of this, it seemed reasonable that terminal substitution by heteroaromatic groups might also lead to rate acceleration of internal hydroamination. To test this hypothesis in the context of the synthesis of (±)-xenovenine (1), the aminodiene 2c was prepared according to the following protocol.
Sequential lithiation of 5-hexen-2-one N,N-dimethylhydrazone 6 (LDA, THF) followed by alkylation with 1,3-dibromo-2-propene (THF, -78 oC ® 0 oC) and subsequent hydrolysis (H3O+) furnished ketone 7b as a mixture of Z- and E- isomers in 94% overall yield. Coupling of 5-ethylthiophene-2-boronic acid (8)5 with 7b [Pd(PPh)4 (5 mol %), Na2CO3, LiCl, 1,2-DME) provided ketone 9 in 66% yield as a 4:1 mixture of Z- and E- geometrical isomers, from which the pure (Z)- isomer could be obtained in 48% isolated yield by chromatography on silica gel. Reductive amination of (Z)- 9 (Na+ BH3CN-, NH4+ -OAc, MeOH) then delivered the requisite aminodiene 2c in 85% isolated yield. Exposure of 2c to complex 3 (10 mol%, toluene-D8, 10 oC) resulted in an efficient and highly diastereoselective monocyclization to generate 2,5-disubstituted pyrrolidine 4c (t/c = 49:1, characterized as the p-toluenesulfonamide) with >95% conversion. It is significant that simply increasing the reaction temperature to 60 oC led to comparatively rapid (18h) and stereospecific bicyclization to afford pyrrolizidine 5c in 90% yield [(NMR), 86% after chromatography]. Reductive desulfurization of 5c (Rainey-Ni, EtOH, 23 oC) ultimately secured (±)-xenovenine (1)6 in 98% yield (44% overall in four consecutive, one vessel transformations) after purification by bulb-to-bulb distillation (Scheme 2).
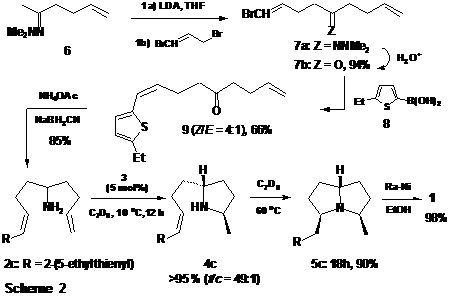 |
.
