AmericanChemicalSociety.com
Reports: B1 45277-B1: Investigation of the Butyllithium-Induced Sigmatropic Rearrangement: Intramolecular Cyclization Reaction of Allyl-Dichlorovinyl Ethers
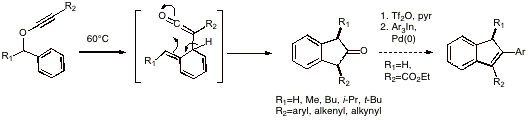
A major goal of our research program is to develop environmentally benign methods for carbon-carbon bond-formation for use in natural products synthesis. Transformations in which multiple bonds are made in a single reaction are of special interest in this context. With the funding we have enjoyed from the ACS Petroleum Research Fund this past year, we have been able to make significant progress along these lines in a number of important areas. In particular, we have uncovered several new and useful transformations of alkynyl ethers. A tandem carbon-carbon bond-forming process, involving [3,3]-sigmatropic rearrangement of substituted benzyl alkynyl ethers, followed by intramolecular 5-exo dig cyclization, gives rise to cis-disubstituted 2-indanones as major products. The larger the benzylic substituent R1 in the starting benzyl alkynyl ether, the greater the syn stereoselectivity (t-Bu>iPr>Me) of the reaction. A remarkable feature of this process is that the sigmatropic rearrangement, which involves the formation of a non-aromatic intermediate, takes place at the relatively low temperature of 60°C; similar aryl allyl ether Claisen rearrangements typically require temperatures in excess of 200°C to proceed! Furthermore, ester-substituted 2-indanones (R2=CO2Et) may by transformed into substituted indenes in high yield by a two-step sequence involving enol triflate formation followed by palladium-catalyzed cross coupling reaction with organoindium reagents or boronic acids.
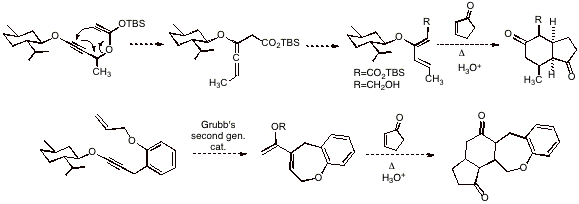
Another sigmatropic rearrangement
process of alkynyl ethers we have been investigating involves the Claisen-like
rearrangement of ester-derived silyl enol ethers; this reaction gives rise to
allenyl ethers which isomerize to donor-acceptor dienes, which may subsequently
engage a range of dienophiles in Diels-Alder reactions to form six-membered
carbocycles. We are also currently exploring the ruthenium-alkylidene catalyzed
metathesis reaction of ynol-ether alkenes, which would provide electron-rich
dienes capable of undergoing Diels-Alder cycloadditions with suitable
dienophiles. This sequential process may allow the formation of up to four
fused rings in a one-pot process, resulting in a rapid increase of molecular
complexity starting from simple acyclic precursors. We anticipate that easily prepared
chiral ynol ethers may allow access to optically active polycyclic products of
relevance in natural products synthesis. Finally, we have initiated the
exploration of gold-catalyzed 6-endo-dig cyclization reactions of
ketone-substituted alkynyl ethers, which would give rise, after treatment with
an alkyllithium or Grignard reagent and hydrolytic quench, to beta-substituted cyclohexenones.
The research outlined above has
had an important impact on the direction of our research program. With the encouraging
results we have obtained, we are now solely focused on the application of these
methodologies in the context of total synthesis.
The undergraduate students who were involved in these projects have
had their first laboratory research experience, and they have gained practical
training in organic synthesis. One of the students is now considering a career
in chemistry and is currently inquiring about graduate level research.