

47257-AC1
Methods for Ligand Scaffold Optimization in Catalysis
This proposal seeks to develop and exploit ligand scaffold optimization as a novel strategy for improving catalyst efficiency. The overall goal of this proposal is to develop methodologies which allow for scaffold optimization and use it to design, prepare, and evaluate focused libraries of new chiral ligands for use in catalytic asymmetric synthesis. The specific aims are to (i) carry out mechanistic studies to improve our understanding of the current generation of self-assembled ligands (SALs), (ii) evaluate new, complementary structural motifs and metal coordination geometries for the preparation self-assembled ligands and catalyst systems, (iii) evaluate the use of boracycles as an alternative motif for self-assembled ligands, and (iv) generate diverse ligand scaffold libraries via click chemistry.
Progress has been made on several of the specific aims. A book chapter (acknowledging support from this grant), was published in Supramolecular Catalysis in which the concept of ligand scaffold optimization was described. Mechanistic studies over the past year (specific aim i), principally via molecular modeling, now suggest that the observed variation in enantioselectivity as a function of scaffold structure is due to the tertiary structure adopted by the ligand scaffold. This differs from traditional chiral catalyst design in which only the ligating group substituents closest to the metal center are thought to create the “chiral pocket” topography; the ligand scaffold serves as a structural element needed to transmit chirality via chiral-relay and provide elements of rigidity. Indeed, the results differ from our original hypothesis that combinatorial scaffold optimization would be used to define only an optimal orientation of the ligating group. The results suggest that the scaffold is part of the chiral pocket topography. Research continues on this aim.
Good progress has also been made on generating diverse ligand scaffold libraries via click chemistry (specific aim iv). A communication has been published in Organic Letters. In it we conclude that click chemistry lends itself to the ligand scaffold optimization approach to chiral catalyst discovery and development. It was successfully used to prepare a small library of chiral triazole diphosphites. While these chiral ligands differ only in the scaffold structure, their performance with respect to yield and asymmetric induction varies significantly in a common test case for asymmetric catalysts, rhodium-catalyzed asymmetric hydrogenation. Enantioselectivity as high as 97% ee is achieved.
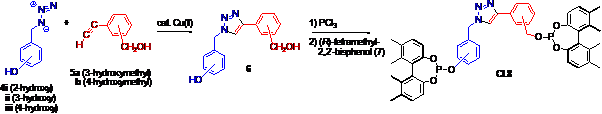
Figure. A facile, modular route to novel ligand scaffolds via click chemistry.
There are four potential ligating sites in CL8 raising questions as to whether it functions as a P,N- or P,P-ligand, or perhaps a tri- or tetradentate ligand, if it indeed chelates at all. Literature data suggests that the P,N-mode is likely. Furthermore, while many macrocyclic metal-ligand chelates have been characterized and others invoked to explain efficient asymmetric catalysis, the 1:1 complex with CL8ib requires a 16-membered P,P-macrocyclic chelate that modeling studies indicate should be quite strained. Nonetheless, other data are consistent with its formation.
First, we find evidence derived from the work of Kagan on non-linear effects in asymmetric catalysis. If the active catalyst is the 1:1 complex, a mixture of enantiomer ligands will give a mixture of enantiomeric 1:1 complexes or, equivalently, a mixture of enantiomeric catalysts and a linear relationship between the observed enantioselectivity of the catalyzed reaction and the enantiomeric purity of the click ligand used is expected; non-linear effects are precluded. The data show a linear response.
NMR and mass spec data are also consistent with the 16-membered P,P-macrocyclic chelate being important to the successful catalysis. The 31P NMR spectrum of the free click ligand CL8ib shows singlets at 130.6 and 135.3 ppm, while its rhodium complex (i.e., (CL8ib)Rh(nbd)(BF4)) shows resonances in 1:1 ratio at 127.0 and 148.1 ppm; each is a doublet of doublets. The coupling constants, JRh,P(1) = 221 Hz, JRh,P(2) = 234 Hz, and JP(1),P(2) = 77 Hz, are consistent with the chelated structure shown. The mass spectrum shows a peak corresponding to the cationic complex. Its isotopic distribution pattern is in good agreement with theory and high resolution mass spectrometry (FAB, 3-NBA matrix) determines the exact mass as 1016.2417 m/z, a value in close agreement with the theoretical value. Thus, 31P NMR and mass spectral analyses, as well as the results obtained using truncated monophosphites and the lack of a non-linear effect, suggest an important role for a 16-membered P,P-macrocyclic (CL8ib)Rh(I) complex in the reaction. A full paper comparing the results obtained using two different ligating groups with a series click-connected ligand scaffolds is in preparation.