44158-AC5
The Structure of Dynamics of Wet Electrons at Metal Oxide Surfaces
Research was performed on the unoccupied electronic states of atoms and molecules on metal surfaces. The unoccupied electronic structure was investigated by experiment and theory. The PRF grant primarily supported the theoretical part of the research by Dr. Jin Zhao.
The GW band structure of TiO2. In the 2007 report, we discussed the electronic structure of O atom vacancy defects on TiO2 surfaces. We extended these studies to the OH impurities, which also reduce the surfaces. Both experiments show a delocalized structure of excess electrons that are introduced by the surface reduction. Our theoretical calculations of the geometrical and electronic structure of reduced surfaces using the GGA method with the PBE functional is in excellent agreement with the experimental structures. Different theoretical approaches that account for self-interaction through inclusion of the Hartree-Fock exchange and correlation, e.g. the B3LYP functional, give localized excess electron distributions and asymmetric distortion of TiO2 lattice that contradict our experiments. Because a significant fraction of the community believes hybrid functionals are superior for describing the electronic structure of metal oxides, we have had difficulty publishing our results. In order to remedy the existing misconceptions on how to calculate the electronic structure of metal oxides, together with Prof. Angel Rubio, we performed a GW calculation of the electronic structure of TiO2. With a perturbation theoretical approach to self-interaction and accounting for the excitonic effects by solving the Bethe-Saltpeter equation, we could reproduce the optical band gap of TiO2 better than is possible by either GGA or B3LYP approaches. In the future, we will extend these calculations to the structure of reduced TiO2 surfaces. Developing a theoretical approach to treating defects in metal oxides, which do not rely on the choice of the functional, will represent a significant advance in our understanding of the electronic structure of these important materials. Publications for the Journal of Chemical Physics and Physical Review B are in preparation.
The m=1 states of alkali atoms on metals. We extended our studies on the electronic structure of
alkali atoms on noble metal surfaces by exploring the unoccupied states derived
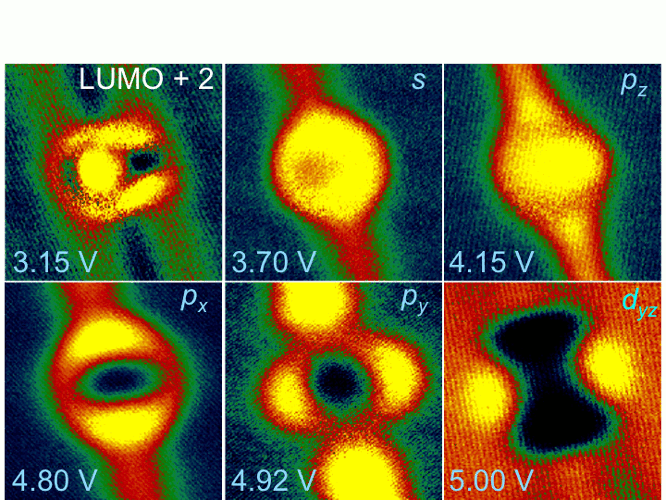
from the px and py orbitals, which have Superatom states of hollow molecules. We discovered a new kind of electronic state that is
particular to hollow molecules such as fullerenes and nanotubes. By spectroscopic imaging of single and
variously aggregated C60 molecules on copper surfaces, we mapped out
the local density of states (LDOS) at different bias energies. In previous STM studies, the LDOS of
the LUMO states with the This research was published in
Science, and will be the subject of a future article in the Accounts in
Chemical Research.
Figure 1. The
dI/dV images of an isolated C60 molecule on copper surface showing
the LDOS of LUMO-2 and s-, p-, and d-SAMO states.