46914-G6
Ab Initio Molecular Dynamics and Ab Initio Quantum Dynamics Investigation of Radical Reactions Involved in Combustion
The original goal of this project was to develop real-time path-integral methods to incorporate nuclear quantum effects into ab initio molecular dynamics simulations. Such methods could be used, for example, to compute reaction rate constants (using on-the-fly electronic structure theory) for small-molecule reactions, including the elementary steps in combustion. While such methods still hold promise, they are extremely computationally demanding, and this prohibits extensive algorithm development using ab initio potentials. At the same time, we are skeptical whether existing versions of density functional theory (DFT), the workhorse electronic structure model for ab initio molecular dynamics, is sufficiently accurate and reliable for such calculations. In particular, self-interaction error in DFT may lead this theory to underestimate reaction barriers. Thus, we have been forced to modify our original goals in order to tackle these problems, but this has led to interesting progress along two new fronts: (1) the development of new model potentials for simulations of aqueous electrons, a task that we originally undertook in order to have a computationally simple (but still scientifically interesting) system on which to test path-integral methodologies; and (2) the development and testing of modified versions of DFT that correct some of present-day DFT's pathologies.
(1) Development of a new model Hamiltonian for the hydrated electron
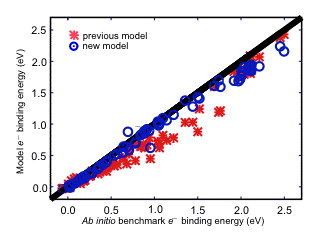
The aqueous electron is an important reactive intermediate in the radiation chemistry of water. Insofar as this system consists of a one-electron wave function solvated by classical water molecules, it has long been a favored testing ground for one-electron quantum mechanics, using some assumed form for the electron-water interaction potential. Although much attention has been paid to parameterization of the electron-water potential, far less attention has been given to the quality of the underlying water model. As part of this project, we have recently parameterized a new electron-water interaction potential, specifically for use with the AMOEBA polarizable water potential. The result is a one-electron model Hamiltonian in which the wave function polarizes the water molecules, and vice versa, in a fully self-consistent fashion. Comparison against ab initio calculations for small (H2O)n- clusters reveals that the new model is significantly more accurate than previous models, both for electron binding energies as well as relative energies of various cluster isomers.
We are currently performing simulations of the bulk aqueous electron, using the new model, in order to understand whether a proper treatment of polarization affects the structure of the bulk aqueous electron. We are also calculating vibrational spectra for clusters in the n = 20-40 size range. In small clusters, vibrational spectroscopy (along with ab initio predictions of vibrational frequencies) has been essential to unraveling the structure of (H2O)n- clusters, and we hope that with our model (which is tractable in clusters with hundreds of water molecules, or in bulk water) will reveal vibrational fingerprints that can be used to understand the evolution of cluster structure as a function of size, which has been an area of controversy between theory and experiment for nearly two decades. Simulations of electronic pump-probe experiments are expected to reveal information about the dynamics of the aqueous electron.
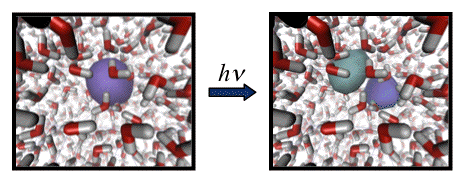
In the long term, this model Hamiltonian will be valuable tool as we work to design efficient algorithms for ab initio path integral molecular dynamics. We are also using this model as a starting point for designing algorithms to perform QM/MM simulations using polarizable force fields for the MM region.
(2) Development of new density functionals
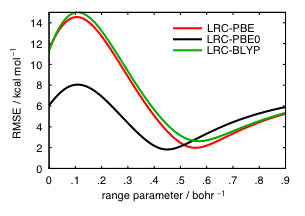
Another point of concern is whether DFT calculations provide barrier heights of sufficient accuracy to be used in rate constant calculations. Much of the error in DFT reaction barrier heights is probably self-interaction error, which can be reduced by application of long-range Hartree-Fock exchange. We have recently shown that long-range-corrected functionals can significantly reduce errors in reaction barrier heights. These functionals (the development of which has also been supported by an NSF CAREER award) turn on the nonlocal Hartree-Fock exchange interaction as a function of one adjustable parameter that partitions the Coulomb operator into long- and short-range components. Our benchmark studies on barrier heights and other properties establish the optimal values of this parameter.