
Back to Table of Contents
46735-G6
Theoretical Investigation of the Two-state Model for the Excess Electron in Saturated Hydrocarbon Liquids Based on Path Integral Simulation
Seogjoo Jang, Queens College of the City University of New York
During
the last funding period, the principal investigator (PI) has completed the
computational code for
the path integral centroid molecular dynamics (CMD) simulation of the excess
electron. This code allows
description of the real time quantum dynamics of the electron and provides
direct understanding of how the dynamics of the electron is correlated with the
local arrangement of surrounding solvent molecules that are also dynamically
evolving. As a test, the
computational code has been implemented for the simulation of an excess
electron in supercritical helium for which well-established interaction
parameters are available.
Figure 1 provides representative samples of the electron (centroid)
trajectories.
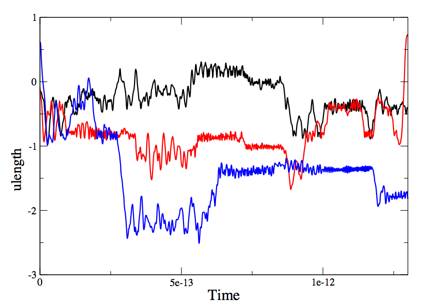
Figure 1. Three representative trajectories (x-components of the
coordinate) of the path integral centroid centroid of the electron. The unit of length is chosen to be ulength=15.3 Angstrom.For
each trajectory in Fig. 1, the electron rattles around a localized region
during most of its time. However,
once in a while (about 0.5 picosecond), it goes through a significant jumping
motion. It is obvious
that the latter motion makes major contribution to the mobility of the
electron. This is
reminiscent of the two-state model for the mobility of the excess electron in
hydrocarbon liquids, and is quite contrary to the widely accepted notion that
the electron in supercritical helium remains localized (in the concentration
tested for the simulation) and is dragged along the motion of helium atoms. Further examination of this issue
will be made for more extensive set of simulation, and a paper reporting the
results will be submitted
during the next funding period.The
above analysis demonstrates the capability of the CMD simulation in revealing
short time real time dynamics that are statistically minor but can make
significant contribution to the mobility of the electron. This demonstrates that the CMD
simulation can be used as a valuable tool for investigating the two-state model
for the mobility of electrons in hydrocarbon liquids. As the first step for such CMD simulations, a computational
program has been developed for the imaginary time path integral simulation of
the excess electron in liquid methane, the simplest hydrocarbon liquid in which
the electron is delocalized substantially. This system represents the other extreme where the electron
is believed to go through coherent quantum dynamical motion most of its time. During the next funding period,
imaginary time path integral simulations will be first performed for this
system in order to get quantitative information on the degree of delocalization
of the electron and its correlation with the nature of the arrangement of
solvent molecules. For
the simulation of the excess electron in larger hydrocarbon liquids, more works
need to be done for the identification of reliable model potentials. A collaborator with good
expertise in the modeling of potential parameters has been sought for since the
beginning of this proposal, and a postdoctoral researcher partly supported by
this grant will start working on this subject in November, 2008. With the development of the model
potentials, path integral simulations for larger hydrocarbon liquids will be
performed by extending that of methane. In
the course of the research, it has become clear that a new quantum dynamics
method that can provide a reliable description of the coherent quantum dynamics
of the electron is needed. The PI
has made significant effort to address this issue, and has been developing a
new theoretical approach combining quantum master equation (QME) formalism with
a polaron transformation.
This approach overcomes the typical weak-system coupling limit of existing QME approaches, and can
establish quantitative understanding of the origin and the effects of the
quantum coherence on the dynamics of electron. An application of a simple version of this theory to a
related problem, the resonance energy transfer of electronic excitation, has
been made in collaboration with other researchers and has been published in the Journal of Chemical Physics as a
Communication in September, 2008.
During the next funding period, the new QME formalism will be extended
to multi-state systems. Then,
application of the multi-state theory will be made to the dynamics of an
electron solvated in samples of hydrocarbon liquids obtained from the snapshots
of path integral simulation.
Incorporation of the QME calculation with the path integral simulation
will provide reliable description of the dynamics of the electron in both
incoherent and coherent regimes, and thus will elucidate whether the two-state
model indeed characterizes its major mechanism of motion in hydrocarbon
liquids.
Back to top
