Reports: ND655476-ND6: Metal-H2 Complexes vs. Metal Hydrides: Nuclear Motion and Implications for Hydrogenation Catalysis
 printer friendly
printer friendlyThis research program examines the quantum delocalization of transition-metal sigma-H2 and hydride complexes—along with complexes intermediate to these two limiting regimes—with the eventual intent of assessing the role of this motion in hydrogenation catalysts. Using quantum chemistry computer simulations, the structural flexibility of these complexes and the role of quantum zero-point energy (ZPE) and tunneling in affecting this flexibility is investigated.
This report will be organized according to the complexes examined. Broadly speaking, the research to-date has focused on the somewhat surprisingly sensitive role of the underlying quantum chemistry methodology. Although density functional theory (DFT) methods will still be used in the path integral molecular dynamics (PIMD) simulations that will yield full-dimensional analyses of quantum motion, determining the correct density functional has proven challenging but surmountable. Historically, precedent on similar transition-metal complexes has guided users of DFT to apply such functionals without benchmarking. As will be shown in the molybdenum complex below, this approach is misguided, at least for these complexes. The low cost of DFT methods is necessarily tempered by the need for highly accurate benchmark calculations that justify the choice of functional. In particular, coupled-cluster calculations with large atom-centered basis sets have been required in the present complexes in order to capture the correct qualitative (and quantitative) nature of the potential energy surfaces.
![]() (Graduate student:
Diana Reese)
(Graduate student:
Diana Reese)
This metal-H2 complex has received the majority of our focus during the report period. Originally inspired by past DFT calculations’ suggestion of multiple low-lying isomers, this complex was shown to adopt a sigma-H2 form, along with both cis- and trans-hydride forms, as shown below:
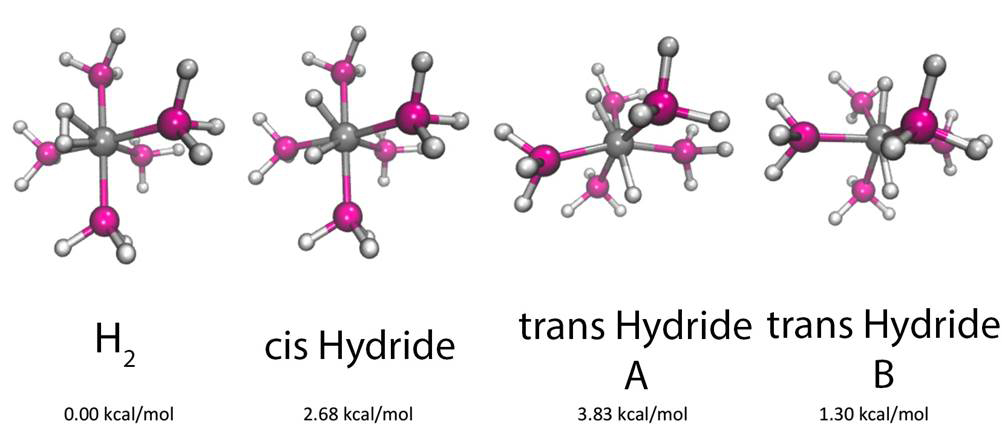
Preliminary calculations, performed in the previous reporting period, suggested that the H2/cis distinction was somewhat blurred by a ZPE-based removal of the connecting barrier. In order to investigate this issue further and complete our understanding of the potential energy surface, H-metal-H potential energy scans were examined with wavefunction-based methods. Due to the challenging nature of the electronic structure of this complex, coupled-cluster methods were required, along with large basis sets. In many cases, single entries in the potential energy curves required weeks of computation time. After careful convergence tests, the CCSD/cc-pVTZ-PP method (with its matched effective core potential) was chosen as a converged reference method. The resulting potential curve along the relevant bending angle is shown below:

Two key results were obtained from this analysis. First, the relative energies of the main isomers are appreciably different from those previously appearing in the literature. The use of accurate wavefunction-based methodology dramatically altered the relative potential energy landscape. Second, the isomer space is larger than previously thought. In particular, the trans isomer can adopt two stable forms, owing to the orientation of the seemingly benign ligands, with one of the trans isomers sitting only roughly 1 kcal/mol above the global minimum.
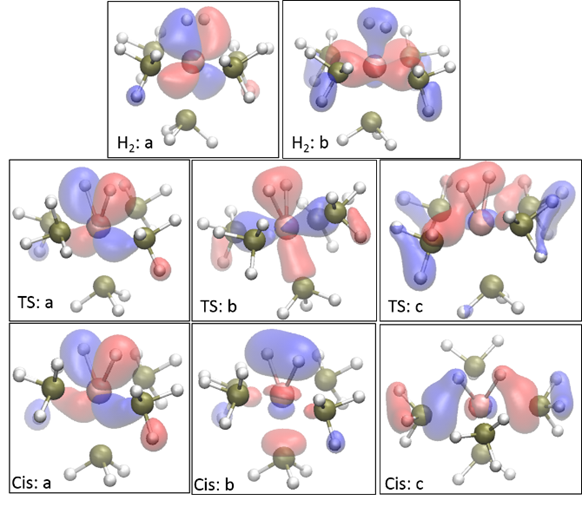 After detailed
investigation of the underlying orbitals, the source of these varying isomers
was also identified. Briefly, the
After detailed
investigation of the underlying orbitals, the source of these varying isomers
was also identified. Briefly, the ![]() orbital
configurations of the metal allow the hydride moieties to be “passed” around
the central metal atom with little energetic penalty:
orbital
configurations of the metal allow the hydride moieties to be “passed” around
the central metal atom with little energetic penalty:
These benchmarks were then used to assess the DFT methods that could potentially be used for path integral sampling. Originally expecting reasonable consistency among functionals, the spread in results was, frankly, shocking. Even with modern density functionals, the qualitative ordering of the isomers could be completely rearranged. Without the accurate benchmarks, the choice of functional would have been pure guesswork. In order to exhaust our options, the more than 100 functionals coded into the Q-Chem software package were analyzed (along with quadrature grid convergence settings), and very few provided reasonable consistency with the benchmarks. The BHHLYP functional, however, provided across-the-board consistency within reasonable error bars, and this functional has been selected for PIMD sampling.
One technical item was addressed during the reporting period, as well. Due to the high cost of even DFT sampling, the barriers involved in this potential energy surface would not be overcome without a prohibitive length of MD trajectory. Instead, the weighted histogram analysis method (WHAM) was added to the PIMD code in order to provide constrained sampling along the previously displayed angular curve. Several unpublished path integral-specific nuances were discovered during this implementation, but all of these technical issues have now been addressed. Sampling is currently underway and will allow for the completion of this study.
![]() (Undergraduate
students: MacKenzie Ferron and Jessica Schulze)
(Undergraduate
students: MacKenzie Ferron and Jessica Schulze)
This complex is of particular interest in this study because it exhibits both dihydrogen and hydride ligands, and interchange of these forms is energetically accessible. The dynamics of this interchange and the potentially non-innocent role of the surrounding ligand motion are questions to be addressed. Furthermore, the role of ZPE in shifting the H2/hydride symmetry breaking will be examined.
In the present reporting period, undergraduates examined the potential energy landscape of these hydride motions. The main findings include the following: Motion should be dominated by dihydrogen motion, consistent with our past studies of purely sigma-H2 complexes. The hydride exchange pathway is higher in energy but accessible on catalytic timescales. This latter pathway does require coupled motion of the dihydrogen and hydride ligands, however. If the ‘outer’ hydrides are considered to be hydrogen donors and acceptors, for example, the donor-acceptor distance must necessarily shrink prior to hydrogen transfer. This off-diagonal cut along the potential energy surface is the key motion involved in hydride exchange.
Using methodology lessons from the molybdenum complex, described above, the benchmarking and functional selection protocols are now well established and can be used to move toward sampling of this complex, as well. Using angular and hydride-transfer cuts, isomer energies and barrier heights will be used to test our suite of functionals. Straightforward extension of the sampling protocol to this complex will then be performed in the remaining funding period.











