Reports: DNI655801-DNI6: Understanding the Effect of Molecular Topology on Surfactants for Enhanced Oil Recovery
 printer friendly
printer friendlyThe goal of this project is to gain a fundamental understanding on the effect of surfactant topology and chemistry on the properties that directly influence its suitability for CO2 flooding applications, including the surfactant's ability to reduce the interfacial tension (IFT) of CO2/water interfaces. This year, we have completed scans of the tail group topology space using a coarse-grained (CG) model based on the SAFT-γ equation of state [Lobanova et al., J. Phys. Chem. B 115, 11154 (2011)] (Figure 1). The main difficulty to obtain the results was finding a consistent criterion to determine the critical surface adsorption (CSA), corresponding to the critical micellar concentration (CMC). We found that the most consistent results were obtained by building histograms of the number of water molecules around the surfactant head groups, with the CSA corresponding to the concentration where the percentage of surfactants with zero or one water unit in the first hydration shell reached 10%. Using this criterion, we ran molecular dynamics simulations for 100 different surfactant designs, obtaining the CSA and surface tension at the CSA for each surfactant. An example showing the results for surfactants containing twelve CG tail group units is shown in Figure 2. We used the results to identify molecular descriptors leading to improved surfactant efficiency and effectiveness, which will be useful to guide experimental efforts to develop new surfactants. A manuscript on this work is currently in preparation and will be submitted during the first quarter of 2018.
Figure 1. An illustration of the CG model surfactant used to predict the effectiveness of different surfactant designs.
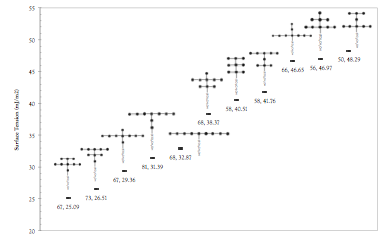
Figure 2. Surface tension at the CSA for surfactant designs containing 12 tail-group coarse-grained units.
In parallel with the calculations using the model of Lobanova et al., we have been developing a model that can be used to consistently build unlike-pair interaction parameters for ionic surfactants to use with the SAFT-γ Mie models. This will enable us to test multiple ionic surfactants without the need for experimental data to find the binary interaction parameters. Instead, the parameters are obtained from ab initio calculations. We extended an approach proposed by Haslam et al., where the pair interactions are estimated from the ionization energy, and multipole moments [Haslam et al., Fluid Phase Equil. 266, 105 (2008)] to be generalized for the Mie potential. We found it necessary to obtain the multipole moments of molecular fragments using the RESP partial charge [Bayly et al., J. Phys. Chem. 97 10269 (1993)] fitting scheme. More specifically, the charges are fit to a MP2/aug-cc-pVDZ electrostatic potential, but using the RESP charge fitting scheme employed by the OPLS force field [Tirado-Rives, J., J. Am. Chem. Soc. 110, 1657(1988)]. In comparison to the standard Berthelot mixing rule for estimating cross interaction parameters, we found our method to be an improvement. In the provided figures we compare the liquid equilibria curve of CO2 with hexane and benzene and the VLE curves between CO2 and water at 383 K. The red lines represent a binary interaction parameter fit to experimental data [Lobanova et al., J. Chem. Thermodynamics 93, 320 (2016)]. Examples showing the predictive ability of this approach for CO2/Hexane and CO2/Benzene mixtures are shown in Figure 3. The results show a clear improvement over the Berthelot mixing rules. Note that this method to estimate interaction parameters is purely predictive, as it uses no experimental data to fit the parameters. We are currently refining our calculations by testing other levels of theory, and finding interaction parameters for molecular fragments that will be used to scan families of ionic surfactants, in analogy with the work on nonionic surfactants.
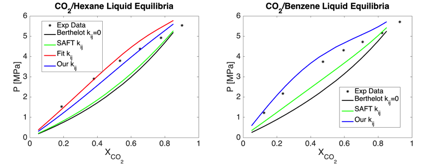
Figure 3. Phase diagrams for CO2/Hexane and CO2/Benzene showing the ability of our approach to predict binary interaction parameters without resorting to experimental data.










