Reports: ND654610-ND6: Mechanisms of Asphaltene Precipitation From Oil: a Multiscale Simulation Study
 printer friendly
printer friendlyDeveloping hybrid simulation approach to asphaltene aggregation
During the third project year, we focused on the final goal of the project: developing of the “association-redistribution” model (ARM) of asphaltene aggregation. We utilize the approach developed in year 2, where the interactions between asphaltene primary aggregates were treated in terms of short range attraction (van der Waals and hydrogen bond) effective described by the Morse potential and longer-range repulsion between the aliphatic sidechains attached to the aromatic cores of the asphaltene and resin molecules. The repulsion creates a barrier for association of the primary clusters (Figure 1), and the height of the barrier depends on the aliphatic chain density.
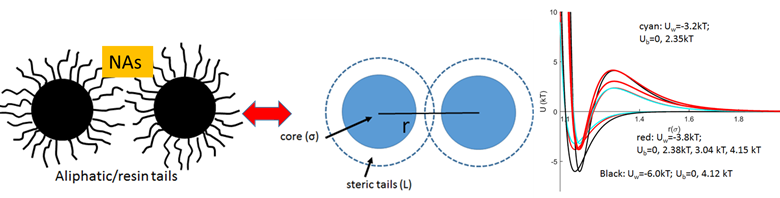
Figure 1. Potential between the primary asphaltene clusters. Aromatic cores interact with Morse potential that reflects van der Waals attraction and hydrogen bonds, while entropic repulsion arises from resins/ aliphatic tails assumed to be rigidly attached on the surface-akin to a functionalized NP and described with de Gennes approach. Overall, the potential has a minimum at short distances, and a steric repulsion barrier has to be overcome for aggregation of two primary clusters. The barrier depends on the surface density of the chains
Since asphaltene and resin molecules in a primary cluster retain a certain degree of mobility, association between two clusters makes a part of the surfaces of both unavailable to the chains and therefore increases the density of the chains on the available surface thus increasing the repulsion between the associated pair and other, non-associated primary clusters. Therefore, we introduced a reaction of a primary cluster association and breakup, with the parameters dependent on the number of neighbors already associated with each cluster. The potentials were analytically derived (Figure 2).
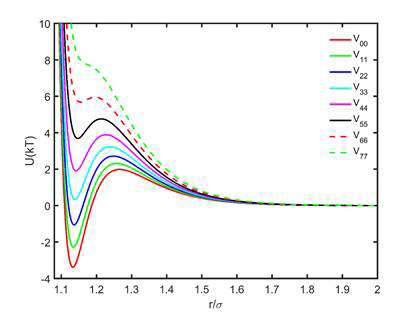
Figure 2. Potential of interaction between two asphaltene primary clusters as a function of the number of associated neighbors (symmetric case, shown on the legend). The more neighbors, the shallower the minimum corresponding to the associated state, and the higher is the barrier that should be overcome to associate.
To model asphaltene aggregation with the ARM model, we devised a hybrid Brownian dynamics / Monte Carlo (BD/MC) scheme that incorporated random association / breakup MC reaction steps into regular BD simulations. The scheme was incorporated into LAMMPS software that allowed simulations of up to several hundred thousand particles. We essentially studied the same system as in the year 2 to explore the effect of taking in account chain redistribution on the colloid structure of asphaltene agglomerates.
In general, the ARM simulations showed that the aliphatic chain redistribution leads to more distinct, long-lasting reaction fractal structures. Typical examples are shown in Figure 3. The system without a barrier showed typical pattern with several regimes (a) very small aggregates have a fractal dimensionality similar to that of diffusion fractals. They are short-living and are not true fractals. The aggregates further agglomerate displaying cluster-cluster aggregation regime. At the same time each agglomerate gradually restructures from a fractal-like structure to a droplet-like structure (middle regime with dimensionality close to 3). The systems modelled with the ARM approach show long-living fractal structures with a single fractal dimension.
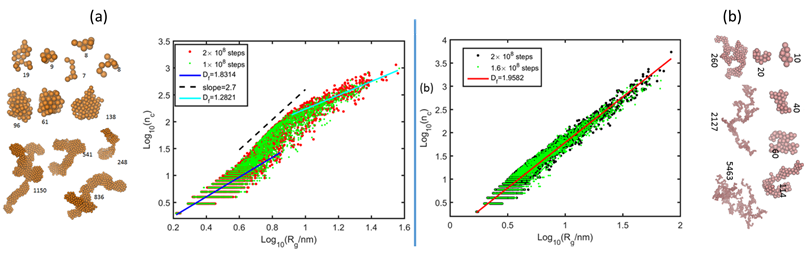
Figure 3. Typical examples of correlations between number of primary clusters in an asphaltene agglomerate and its radius of gyration for a system with low and association-independent potential (a) and with the ARM model (b)
Overall, we developed a new modeling approach to larger scale simulations of systems of associating particles that are applicable far beyond the specific examples of primary asphaltene clusters. Similar situation may be, for example, found in systems of nanoparticles onto which polymer chains are adsorbed. Using the new ARM model, we demonstrated the importance of correct accounting for the steric entropic repulsion between the primary clusters and bigger agglomerates that depend on their structure. The results are prepared for publications in peer-review journals.
Impact on students involved in the project.
The project involved two graduate students (Tianying Ma and Ravish Kumar), both of whom graduated who graduated with Master degrees in Chemical engineering, two undergraduate students (David Woo and Nish Basheri) and a postdoc (Santo Polouse). All students and the postdoc received one-on-one instruction from the PI. Working on the project not only give them experience in programming and modeling techniques. Undergraduate student David Woo (who took 3 research credits) presented his work at the Rutgers School of Engineering undergraduate symposium and was awarded the best poster award by the faculty. Since than he has graduated and was hired by Collagen Matrix Inc as a research technician. Nish Basheri, an undergraduate student, who recently started working with the PI, received an Undergraduate Research Assistance fellowship from Aresti foundation; the topic of his project is “Nanostructure and dynamics of heavy fractions of crude oil” and his undergraduate research was supported for 2016-2017 academic year.










