Reports: ND656673-ND6: Accurate and Predictive Petroleum Molecular Modeling
 printer friendly
printer friendlyThere is a growing need for predictive molecular simulations and next-generation force fields in the energy industry. The goal of this project is the development, implementation and evaluation of next-generation classical force fields designed for use in energy-relevant simulations in diverse environments, including high pressures and temperatures. In the first year of this project classical potentials for the C2 hydrocarbons (ethane, ethylene, and acetylene) were developed and work on atom typing and fitting dispersion coefficients for a larger set of energy-relevant molecules, setting the stage for a more exhaustive force field, has been completed. The importance of this work includes evaluating the performance of different repulsion/dispersion potentials, exploring different fitting methods for classical force fields and evaluating the importance of many-body interactions, including explicit polarization and many-body Van der Waals.
The C2 hydrocarbons were chosen because of their importance to the petroleum industry while retaining simplicity and computational tractability. These models were developed through the following methodology based on previous considerations and constant reevaluation. The equilibrium geometry were obtained from a geometry optimization at the CCSD(T)/aug-cc-pvqz level of theory with the electronic structure program ORCA. Partial charges, located on atom-centered sites, were obtained using the CHELPG method of fitting the classical electrostatic potential to that of the orbital optimized CCSD/aug-cc-pvqz electronic density as implemented in ORCA. Atom-centered static polarizabilities were obtained with the default procedure implemented by the CamCASP program. Briefly, the molecular orbitals of a gas phase monomer were calculated at the PBE0/aug-cc-pvqz level of theory with the CS00 asymptotic correction as implemented in NWChem. The total molecular polarizability and point to point polarizabilities between 2,000 points randomly located on the 2x-4x van der Waal surface were then calculated with the CKS propagator. These molecular polarizabilities were transformed to atom-centered point polarizabilities using the localization procedure of Le Sueur and Stone (1994). For models including the asymptotically correct dispersion coefficients these atom-centered point polarizabilities were integrated over the imaginary frequency domain.
Several different families of models were developed for the purposes of comparison and compatibility with existing forcefields. These include models based alternatively on Lennard-Jones repulsion/dispersion as well a form incorporating the three leading asymptotically correct dispersion coefficients and exponential repulsion to more realistically model repulsion/dispersion interactions. The remaining parameters (either Lennard-Jones or exponential repulsion) were fit to ab initio single point energies of 2,000 randomly generated trimer configurations containing all possible combinations of C2 hydrocarbons. The energies were computed at the DLPNO-CCSD(T) level of theory utilizing a aug-cc-pvdz/aug-cc-pvtz extrapolation to the complete basis set limit and basis set superposition error corrected by the counterpoise method using ORCA. The goodness-of-fit is shown for the various models in Figure 1 and demonstrate the need for more realistic modeling of repulsion/dispersion interactions – a point that is often underestimated in extant work in this area.
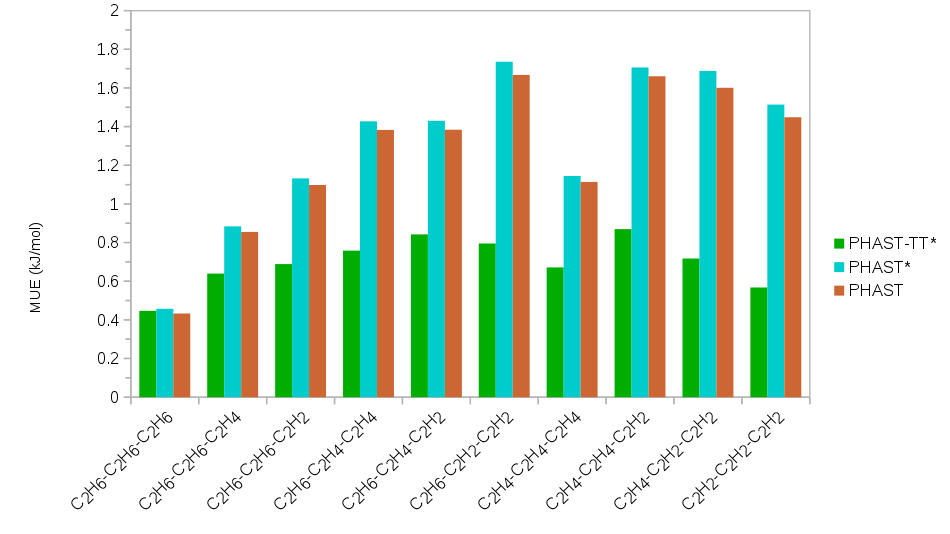
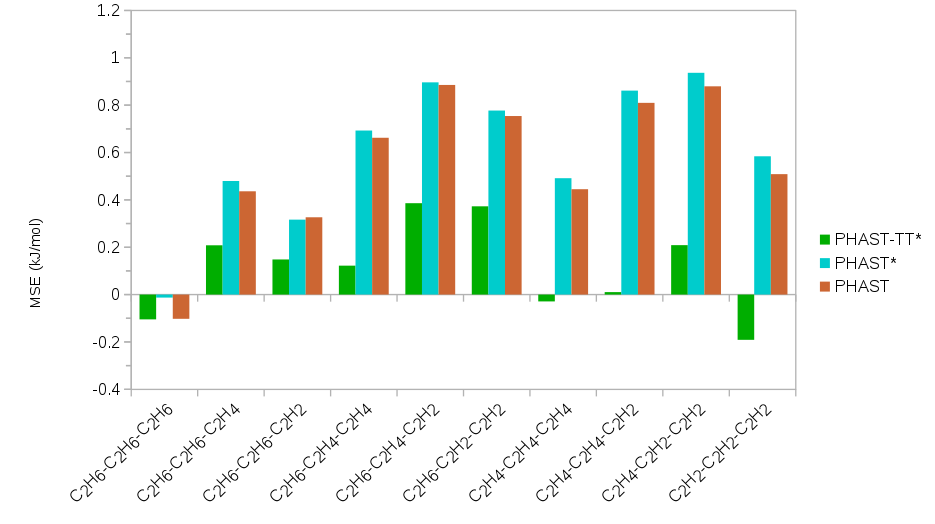
Figure 1. The mean unsigned error (MUE, above) and mean signed error (MSE, below) of the fits to DLPNO-CCSD(T) in kJ/mol. PHAST-TT* indicates the models whose repulsion/dispersion is represented by Tang-Toennies damping of the three leading asymptotically correct dispersion coefficients and exponential repulsion while PHAST and PHAST* indicate models whose repulsion/dispersion is based upon the Lennard-Jones equation. An asterisk (*) indicates explicit polarization is included in the model.
Work is in progress on fitting similar force fields for a larger set of energy-relevant compounds including larger compounds and those with diverse functional groups. In the interest of a more universal and transferable force field work is continuing with the notion of atom typing, i.e. constraining parameters for atoms in a similar chemical environments together. The asymptotically correct dispersion coefficients for this set of compounds have been fit in an iterative manner wherein the dispersion coefficients of each atom type are optimized in a manner similar to the C2 hydrocarbons with the dispersion coefficients on all other atom types held constant. These new dispersion coefficients are then carried over to the next iteration and this process is carried out to self-consistency. This naturally leads to the ability to discern when new atom types might be needed as well to investigate transferability with regards to atom types in general. The results of the fit are illustrated in Figure 1 and demonstrate the need for this kind of iterative process to correctly parameterize polarizabilities and dispersion coefficients.
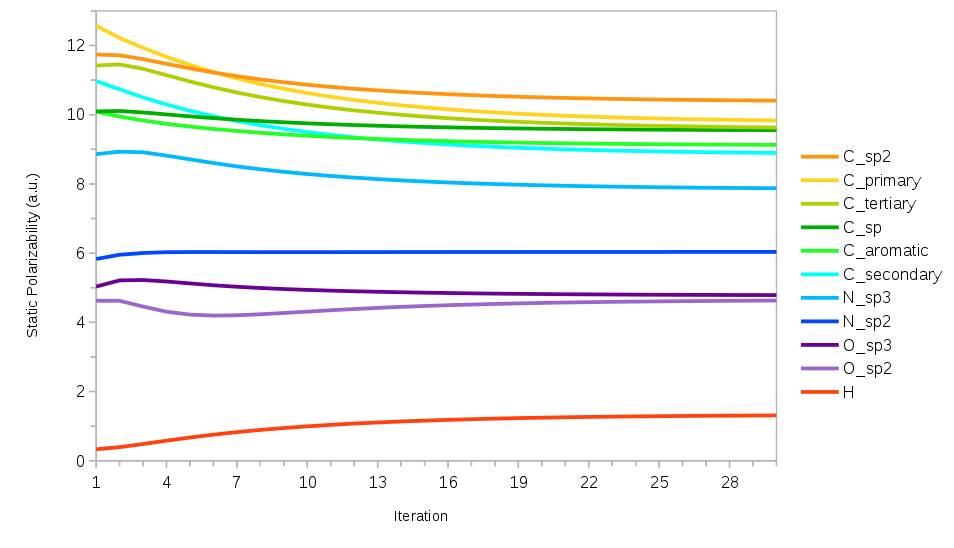
Figure 2. The static dipole polarizability (in a.u.) as a function of the number of iterations in the dispersion coefficient fitting process for the larger set of energy-relevant compounds and the set of atom types considered.
This grant has supported one graduate student, Adam Hogan, and one undergraduate student, Zac Dyott. Adam Hogan is currently on his fifth year of graduate school and has a manuscript resulting from this research in preparation. Zac Dyott graduated in the Spring 2017 semester and is now a first year graduate student at the University of Wisconsin–Madison.










