Reports: DNI156159-DNI1: Photocatalytic C-F Functionalization: Transforming Perfluoroarenes from Inert Molecules to Synthetic Linchpins
 printer friendly
printer friendlyThe impact of this award is significant and its impact will continue to grow in importance with more time. With this award, an undergraduate researcher, Ryne Overfield, was paid over the summer to perform research. As a result of his experience, he chose to stay on for graduate school at OSU. While not yet certain, it appears likely that he will join my research program. For the PI, tenure was granted a full year prior to the normal time frame. Receiving the ACS PRF-DNI was taken as an external validation of the value of the PI’s research program and merit of his science and certainly contributed to the early tenure decision. Additionally, the Weaver lab was able to add a post-doc to the group. However, with respect to this, the impact has been somewhat limited. The search and hire was delayed significantly last fall due to the uncertainty in changes to the federal minimum wage of the post-docs. However, this was resolved by the beginning of this calendar year and the post-doc hired. However, due to prior obligations, the post-doc was unable to commence work until June 2017. Three months later he took a permanent research position at a company. So between delay in the hiring, starting, and premature departure of the post-doc, the project has not advanced as much as desired. This will be resolved as new personnel come on board.
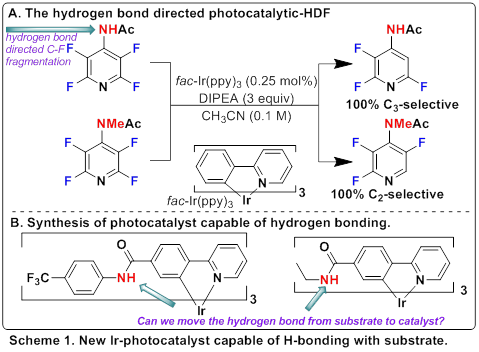
The Weaver group has developed a number of chemical reactions which involve photocatalytic electron transfer to a perfluoroarene which results in an unstable perfluoroaryl radical anion. This radical anion undergoes fluoride fragmentation to give a perfluoroaryl radical. In these chemical reactions we have investigated how to utilize the perfluoroaryl radical for synthetically useful chemistry. In other words, we have investigated how the radical could be used to form new C–C bonds. In addition, we have begun to learn what controls the regioselectivity of the fluoride fragmentation. There are often as many as five different fluorines and in almost all cases the reactions are highly selective in the fragmentation event. However, to try to advance the chemistry further, we are trying to understand what factors control this selectivity, and how we can manipulate it. We hypothesized that one important feature is the deviation from planarity of the radical anion. In the perfluoroaryl radical anion, the molecule is no longer planar or aromatic, the fluorides bend out of the plane of the ring. Based on the literature, the fluoride that deviates the most from planarity is the one which undergoes fragmentation. This is dictated by the electronics of the substrate. We speculated that we might be able to use non-covalent interactions to cause a fluoride not normally prone to fragmentation, to undergo fragmentation. Indeed, we showed that a strategically located intramolecular hydrogen bonding interaction, could be used to completely overcome the normal C–F regioselectivity (Scheme 1A). The objective of this project has been to build in these types of regiochemical altering, non-covalent interactions into the photocatalyst, rather than the substrate. This could potentially allow alternative regiochemical patterns to be dictated by the choice of photocatalyst, rather than being limited to substrate controlled fragmentation.
The synthesis of the appropriate photocatalysts is challenging, but we have synthesized and characterized a couple of novel photocatalysts (Scheme 1B). In our first experiments, we were excited to see a dramatic departure from the normal reaction outcome with a model substrate. This result suggests that this strategy may be successful in opening up new regiochemcial outcomes in the photocatalytic C–F functionalization. Ultimately, this would give rise to higher value organic molecules, which are themselves derived from petroleum feedstocks.
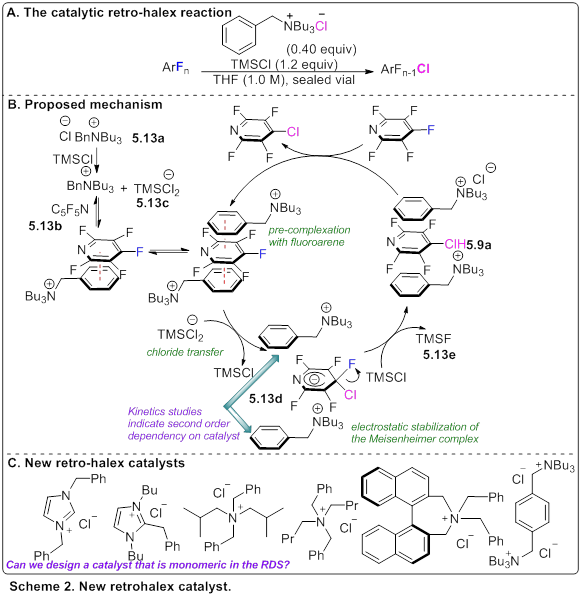
In another approach, we have begun to design catalysts with another type of non-covalent interaction that can provide alternative reactivity or selectivity during C–F functionalization, we have observed that BnNBu3 can serve as an efficient and selective catalyst for the retro-halex reaction (Scheme 2A). Specifically, it can catalyze the conversion of C–F bond to give a C–Cl bond, under the right conditions. During our studies of this process we determined that a donor-acceptor complex between the benzyl of the ammonium and the electron deficient perfluoroarene was a key design feature. We hypothesized that this donor-acceptor interaction served to bring the ammonium and the perfluoroarene close to one another, which would otherwise be electrostatically repelled from one another. This in turn, brought the anion, TMSCl2-, into close proximity and minimized the atomic movement necessary in order to stabilize the key Miesenheimer intermediate (Scheme 2B). Kinetic studies indicated that in the rate-determining step two catalysts were involved. As a consequence, relatively high catalyst loadings (ca. 40 mol%) were required to achieve reasonable reaction rates. We hypothesized that the synthesis of a single molecule which contained the key stabilizing components in the appropriate orientation, could result in significantly enhance kinetics, and potentially an expanded scope. We began designing new catalysts which contained multiple potential stabilizing or attractive features within a single molecule (Scheme 2C). Early kinetic experiments suggests that the imidazolium catalysts are monomeric in the RDS, though they are less efficient.