Reports: ND655534-ND6: Chemistry by Fiat: Quantum Biased Molecular Dynamics
 printer friendly
printer friendlyThe nature of the hydrated electron, which is an excess electron embedded in liquid water, continues to be a source of much debate. The consensus view until fairly recently was that the electron locally expels the water and primarily occupies a cavity. Recently, however, our group challenged the cavity picture, and based on mixed quantum/classical simulations with a rigorously derived electron-water interaction potential, we suggested that the hydrated electron occupies a region of enhanced water density with little to no appreciable central cavity. All-electron calculations based on density functional theory (DFT) also paint a picture in which there is significant overlap of the excess electron's wave function with the surrounding water, albeit with a small central cavity. The problem is that most DFT calculations do not use a functional that is appropriate for describing the electronic structure of the water as well as that of an excess electron that resides essentially entirely between the water molecules.
With our recently-developed quantum umbrella sampling algorithm in hand, we have turned toward DFT-based simulations of the hydrated electron. To improve on the DFT-based work that has been performed previously, our goal is to perform simulations with the range-separated hybrid BNL functional, combined with the D3 dispersion correction, which although computationally expensive should be the most accurate for simulating this particular system. Moreover, rather than running fixed-point calculations on selected gas-phase water anion cluster geometries or small clusters carved out of mixed quantum/classical trajectories, our goal is to run actual ab initio molecular dynamics simulations. Our estimate is that we can readily do anion clusters with 20 waters, and 32 waters should be within computational reach. With our umbrella sampling, we can then compare the nature of internally-bound versus surface-bound clusters, as well as clusters where electron is forced to be more or less cavity-like.
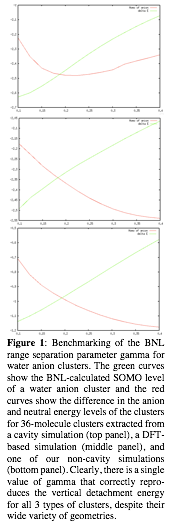
One issue with density functional theory-based calculations to date is that no one has carefully benchmarked any functional with the goal of being able to correctly simulate both pure water and water with an excess electron. Most hybrid functionals have a lot of self-interaction error, and of course dispersion needs to be explicitly accounted for, as documented in the citation mentioned elsewhere in this report. Also ignored in the water DFT community is making sure the theory correctly describes the band gap of liquid water: if one wishes to simulate hydrated electrons and compare the calculated energy levels to those measured in photoelectron spectroscopy experiments, it's important that the energy levels of the water are correct. To remedy this situation, we have taken advantage of Danny Neuhauser's (a UCLA colleague of the PI's) range-separated hybrid BNL functional (along with the D3 dispersion correction). BNL, although more computationally expensive than other functionals to implement, has been shown repeatedly in the literature to produce the best energetics compared to high-level calcuations for a variety of situations, including where weak or charge-transfer interactions are the dominant contributions. We already have shown that BNL does an excellent job reproducting both the energies and band gaps of neutral water clusters, and in the past few months we have begun optimizing the range tuning parameter gamma for the simulation of water anion clusters. To optimize gamma, we pulled configurations of the 36 closest water molecules to the electron's center of mass from various one-electron calculations, including the standard cavity model, our non-cavity model, and previous 'hybrid' models that are based on a different DFT functional than the BNL functional used here. What we find is that there is indeed a single value of gamma that can correctly reproduce the vertical detachment energy of all three types of water anion clusters, as shown in Figure 1. We also have benchmarked against several dozen 6-water molecule anion clusters for which the energies are known at the CCSD(T) level, and again found that the same value of gamma provides the best agreement. With this value in hand, we are currently running MD simulations of room-temperature clusters with 6, 11 and 20 water molecules.
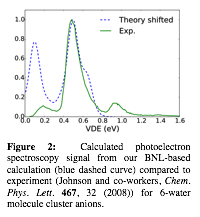
We plan on then quenching uncorrelated configurations from these trajectories and comparing the VDE's to various photoelectron spectroscopy experiments, as well as comparing the structures to the predictions of cavity and non-cavity pseudopotential calculations. Figure 2 shows preliminary results for the calculated photoelectron spectroscopy of 6-molecule water anion clusters. The calculations predict that there are two significant binding motifs (blue dashed curve), with vertical detachment energies of ~0.1 and ~0.5 eV, which are in excellent agreement with what was observed experimentally by Johnson and co-workers (Chem. Phys. Lett. 467, 32 (2008), green curve). The trajectories with 11 and 20 waters are still running, but all indications are that there will be similar excellent agreement between the simulations and the experiments. Interestingly, without any umbrella potential, all of the trajectories for all water cluster sizes show the electron on the surface of the cluster and not the interior; the multiple binding motifs appear to originate form configurations where exterior waters make either a single H-bond (weak binding) or two H-bonds (strong binding) to the excess electron.
Once these base trajectories are complete, we will apply are umbrella sampling to explore the full nature and relative free energies of the different binding motifs of the excess electron to water anion clusters.
| Figure 1: Benchmarking of the BNL range separation parameter gamma for water anion clusters. The green curves show the BNL-calculated SOMO level of a water anion cluster and the red curves show the difference in the anion and neutral energy levels of the clusters for 36-molecule clusters extracted from a cavity simulation (top panel), a DFT-based simulation (middle panel), and one of our non-cavity simulations (bottom panel). Clearly, there is a single value of gamma that correctly repro_duces the vertical detachment energy for all 3 types of clusters, despite their wide variety of geometries.
|
| Figure 2: Calculated photoelectron spectroscopy signal from our BNL-based calculation (blue dashed curve) compared to experiment (Johnson and co-workers, Chem. Phys. Lett. 467, 32 (2008)) for 6-water molecule cluster anions.
|