Reports: ND455838-ND4: Effect of Polarity Reversal on Rates of Hydrogen Atom Transfer Reactions
 printer friendly
printer friendlyOur work on the resolution of asymmetrically substituted cyclopentadienyl ruthenium complexes was summarized in last year's report.
We have concluded that it is not practical to do enantioselective hydrogen atom transfers from transition-metal hydrides, and have shifted our attention to other fundamental questions about that reaction. In particular, we have examined the effect of polarity reversal on the rates of such reactions.
In 1999 Roberts1 reviewed the effects of the electronegativity of A and B on the rate of a hydrogen atom transfer between them. He defined as "electrophilic" radicals with a high ionization energy and a high electron affinity, and defined as "nucleophilic" radicals with a relatively low ionization energy and electron affinity. He proposed that H• transfer would be faster between radicals of opposite polarity than between radicals of the same polarity (below).
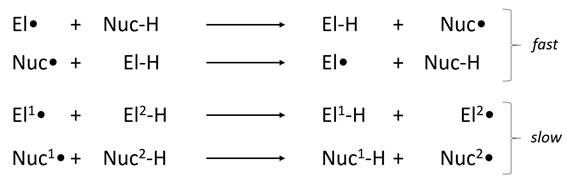
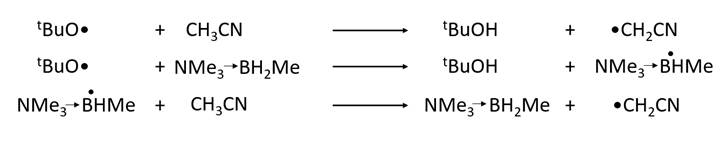
The transfer of H• from CH3CN to a t-butoxy radical is for example relatively slow — both tBuO• and •CH2CN are electrophilic. The transfer can be carried out much more quickly if it is done in two steps. For example, transfer to tBuO• from the boryl radical below (H• abstraction by tBuO• from Me3N®BH2Me) is fast because the boryl radical is nucleophilic and the transfer thus involves polarity reversal (transfer from a nucleophilic radical to an electrophilic one). Transfer from CH3CN to the boryl radical (also below) is also fast because it also involves polarity reversal.
We have wondered how these principles affect H• transfer to and from transition-metal radicals. Transition metals are in general electropositive, so their metalloradicals should be nucleophilic and their transfer of H• to many carbon radicals should be "slow". We have looked at, for example, the reaction of (C5Ph5)Cr(CO)3H with Ar3C•, where Ar = p-tBuC6H4. (The tBu substituents block the head-to-tail dimerization characteristic of other trityl radicals.) Such trityl radicals are easily oxidized, and thus can be classified as "nucleophilic".
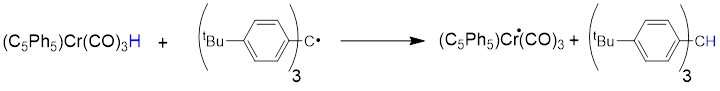
The reaction above is indeed slow, easily monitored on a UV-vis spectrometer by following the absorbance of the trityl radical at 523 nm. The second-order rate constant is only 0.013 M–1s–1 at room temperature.
H• transfer from a transition-metal hydride like (C5Ph5)Cr(CO)3H to an electrophilic radical like TEMPO should be fast. Indeed, the second-order rate constant from Figure 1 below is 139.5 M–1s–1 at room temperature.
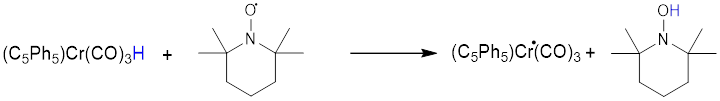
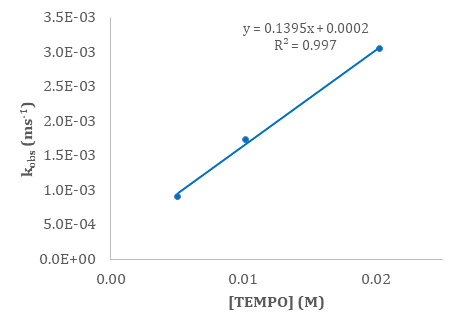
Figure 1. kobs (ms-1) versus excess concentration of TEMPO for H• transfer from (C5Ph5)Cr(CO)3H to TEMPO.
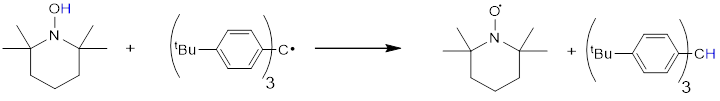
We have also measured (again by UV-Vis) the rate constant for the other step with polarity reversal (H• transfer from TEMPOH to Ar3C•, below). The rate constant at room temperature is 0.04 M-1s-1.
With a kinetic simulation program we have been able to calculate the effect of 10% TEMPO on H• transfer from (C5Ph5)Cr(CO)3H to Ar3C•. As shown in Figure 2, some catalysis should occur.
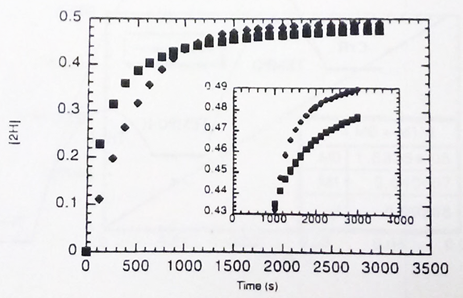
Figure 2. Simulation of catalyzed and uncatalyzed H• transfer from (C5Ph5)Cr(CO)3H to Ar3C•.










