Reports: ND554130-ND5: Design, Processing, and Characterization of Advanced Binary Nanoparticle Catalysts
 printer friendly
printer friendlyThe ACS ‘New Directions’ grant has catalyzed several new research endeavors directed at photochemical and electrochemical CO2 reduction in our laboratory. The photochemical efforts described in previous reports form the basis for our participation in the carbon X prize. This report describes out recent work on electrochemical CO2 reduction.
Electrochemical CO2 reduction reaction (CO2RR) has garnered strong interest as a promising pathway to convert CO2 emissions into higher value chemicals including fuels and hydrocarbon feedstocks. In particular, copper has been shown to produce a range of useful hydrocarbons at room temperature, which could be powered renewably. The main challenges involved with CO2RR is the low selectivity to one product, competition with the hydrogen evolution reaction (HER) and the overall high overpotentials required. A recent ACS Catalysis report by Kumar et al. (DOI: 10.1021/acscatal.6b00857) showed that pulsing the electrochemical potential during reduction increased H2 and CO selectivity. Intrigued by this technique we sought out to understand the underlying mechanism and expand on this initial probe.
We used a standard three electrode setup, following procedures established by previous electrochemical CO2RR studies. Figure 1 shows a faradaic efficiency (FE) ‘baseline’ for our copper working electrode under constant potential, which is consistent with previous studies. We analyzed the composition of reaction products via gas chromatography (GC) and high performance liquid chromatography (HPLC). We focused on detecting H2, CO, CH4 and C2H4, and HCOOH, as these major products are known to compose ~90-98% of the FE.
A square-wave potential was modulated with varying pulse times to examine how pulse duration affects the CO2RR selectivity. Using a 0.1 cm2 copper electrode we kept the anodic potential ceiling (Ea) at 0 V vs. Ag/AgCl (+0.6 V vs. RHE) and varied the reduction potential ceiling (Ec). The cathodic and anodic pulse intervals were kept symmetric. The resulting product distributions were compared to the baseline FEs. Figure 1b-d shows the FE for pulse intervals of 1 s, 100 ms, and 50 ms, respectively. Remarkably, HER is suppressed at all measured potentials more negative than -1 V vs. RHE for the 1 s pulse and all measured potentials for the millisecond pulse intervals. Furthermore, CH4 is selectively favored for all pulse intervals, especially at measured potentials more negative than -1.0 V vs. RHE. For the 50 ms pulse CH4 FE dominates throughout the potential window. The selectivity for CO is favored in comparison to the baseline for less negative potentials (e.g, at 100 ms and -0.85 V vs. RHE).
We hypothesize that the observed product selectivity is affected by the dynamic surface proton coverage of the catalyst during potential pulsing, as shown in Figure 2. It is well-accepted that the surface affinity of CO determines the reduction of CO2 and product selectivity. During the cycle of the anodic pulse at +0.6 V vs. RHE, the electro-adsorbed proton can preferentially electro-desorb (Had → H+ + e–) compared to other intermediates. This drives the hypothesis that surface proton coverage is reduced through each anodic pulse. As surface protons are known to be a HER intermediate, diminished coverage would suppress the HER. Additionally, CO has been shown to adsorb during the anodic potential, therefore the pulsed electrode will preferentially accumulate CO2 intermediates over hydrogen as depicted in Figure 3.
We found that the application of a pulsed potential allows us to substantially suppress the HER while shifting selectivity to CH4 and CO. Millisecond pulse intervals at highly negative potentials favored CH4, while short pulse intervals at less negative potentials favored CO. We attribute the improved CO2RR selectivity to a re-arrangement of surface hydrogen coverage during the pulsing. We also establish that the size and geometry of the electrode matters; when using a small copper electrode, HER was suppressed to less than 5% and methane or CO could be selectivity produced even at fairly low potentials. We found that the electrode size, which dictated the response time of the pulsing current, determined whether our results or Kumar’s results were achieved.
On a fundamental level, our findings provide new insights into the timescales of competing pathways in CO2RR. From an applied perspective, our results present an opportunity for the product selectivity to be tuned by adjusting the temporal profile of the electrochemical potential. We anticipate our findings may help others to further understand the CO2RR pathways and improve performance for other catalysts.
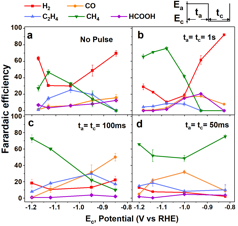
Figure 1. Faradaic efficiency for various products as a function of cathode ceiling potential. (a) constant potential. (b) pulsed potential 1 s on, 1 s off. (c) pulsed potential 100 ms on, 100 ms off. (d) pulsed potential 50 ms on, 50 ms off. All results were measured using the anode ceiling potential of +0.6 V vs. RHE.
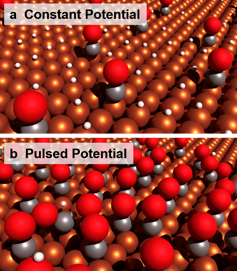
Figure 2. Conceptual depiction of adsorbed species on the copper electrode surface. (a) High proton surface passivation during a constant reduction potential. (b) High CO surface passivation during a pulsed potential.
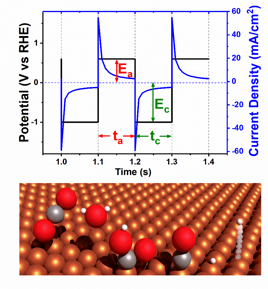
Figure 3.










