Reports: DNI156372-DNI1: Titanium-Mediated Selective Dihalogenation for the Vicinal Difunctionalization of Simple Alkenes
 printer friendly
printer friendlyRecent advances in our laboratory have enabled the regio- and enatatioselective dihalogenation of a wide range of allylic alcohols using a chiral Schiff-base as the catalyst.[1] With these new methodologies developed in our laboratory we now have access to a wide range of dihalogenated motifs and the ability to study these building blocks from a synthetic chemistry standpoint. We have been investigating three distinct platforms for the functionalization of dihalides, all of which involve the generation of a halonium ion followed by subsequent nucleophilic capture. Firstly, a halophilic metal, such as Ag(I), could be utilized for the generation of a halonium and the metal counterion could serve as the nucleophile for its capture. Secondly, inspired by Reetz’s results, we propose to investigate the reactivity profile of a variety of organometallic compounds with dihalides with the aim of discovering yet unidentified reactivity. Thirdly, inspired by Denmark’s study, it is possible that simply the solvent, if ionizing enough, could heterolytically dissociate one of the C–X bonds to form a halide anion and a halonium, and this reactive intermediate would then be captured by a nucleophile in solution. As well as accessing the halonium intermediate in different ways, we also aim to investigate a wider range of nucleophiles than the ones traditionally used (Scheme 1).
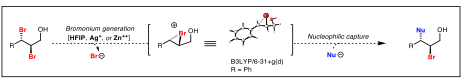
Scheme 1. Research design. Bromonium ions can be generated from dihalides and subsequently captured by a variety of nucleophiles.
Silver(I) can readily generate haloniums from vicinal dihalides with subsequent nucleophilic substitution by the silver counterion. However, due to lack of access to enantienriched dihalides, the enantiospecificity of these processes have never been investigated prior to our work. Our laboratory found that silver salts (AgF, AgOBz and AgOAc) allow for stereoretentive substitution at the benzylic position of dibrominatied cinnamyl alcohol, with nearly perfect enantiospecificity, presumably via a configurationally stable bromonium (Scheme 2A). 1a
Silver salts were also investigated in combination with electron rich arenes to allow for Friedel-Crafts type reactivity between bromoniums and aromatic π-systems. Treating dibromide with AgOTf in a solution of benzene gives rise to substitution of the benzylic bromide with the phenyl ring in moderate yield. Reacting the same dibromide with AgSbF6 and 1,3-benzodioxole provides the analogous product in 40% yield (Scheme 2B).
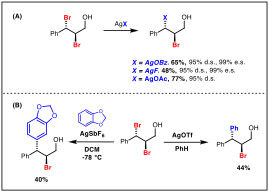
Scheme 2. Silver-mediated functionalization. (A) Substitution by silver counterion. (B) Friedel-Crafts type reactivity.
Other dibromide substrates were also subjected to the same reaction conditions. We found that non-benzylic secondary halides react very slowly, but AgF did produce low yield of bromofluoride products (Figure 1). Additionally, substrates that contain a tertiary halide are prone to undergo elimination pathways especially in the presence of basic counterions yielding a variety of aldehyde and allylic bromide products.
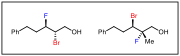
Figure 1. Other accessible products with silver.
We proposed that if a dihalide was reacted with an organometallic species, the halophilic metal could abstract a halide and generate a bromonium ion, with subsequent nucleophilic capture by the C–[M] bond. We have found that dialkyl- and diarylzinc species readily undergo substitution reactions with cinnamyl dibromides forging alkylated or arylated products at the benzylic position without any alkyl shifts observed (Scheme 3A). The pendant alcohol functionality is crucial for success of this transformation. We propose that zinc might coordinate to the deprotonated alcohol and the alkyl/aryl group is delivered intramolecularly to the bromonium.
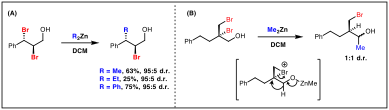
Scheme 3. Reactions with dialkyl- and diarylzinc reagents.
When using another dibromide starting material, a different reaction pathway is operative involving a proposed hydride shift from a bromonium intermediate, followed by nucleophilic addition to the transiently formed aldehyde yielding a diastereomeric mixture of secondary alcohols (Scheme 3B). Further work will involve improving the efficiency of this reaction and fully exploring the nucleophile and dihalide scope.
We envisioned that if an external nucleophile is present in a strongly polar solution, an ionization pathway could be intercepted for a productive bond-forming reaction. When dissolving cinnamyl dibromide in HFIP/water, the anti-bromohydrin product forms rapidly in high yield. Other nucleophiles, such as fluoride (CsF), azide (NaN3), hydride (Et3SiH) or a phenolate (Na-creosolate) can also be incorporated (Scheme 4). The basic nature of some of these nucleophiles can lead to unwanted side reactions, but further optimization will aim to identify reaction pathways, such as buffered medium, where these side reactions are suppressed.
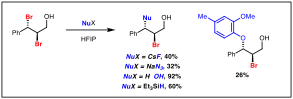
Scheme 4. Forming carbon–heteroatom bonds in HFIP.
Notably, this method can be extended to the addition of carbon-based nucleophiles. Analogous to Friedel-Crafts alkylations, electron-rich arenes can intercept the bromonium ion to form a product with a new benzylic carbon–carbon bond. Toluene, 1,3-dimethoxybenzene, and 1,3-benzodioxole are all capable nucleophiles in this reaction. Further investigation has shown that sp3–sp3 carbon–carbon bonds can be formed using allyltrimethylsilane as the nucleophile (Figure 2).
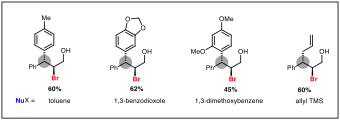
Figure 2. Formation of C–C bonds in HFIP.
Extending this method for non-benzylic dihalide substrates proves somewhat challenging (Figure 3). These undergo slower solvolysis, requiring extended reaction times and heating. Additionally, elimination pathways are observed, possibly through the intermediacy of bromonium ions. We are hoping to identify strategies that circumvent these problems. For instance, using iodochlorides in place of dibromides increases reaction rates with a concomitant drop in aldehyde formation. A strategy to avoid the formation of elimination side products would be to render these processes intramolecular so that nucleophilic capture could outcompete elimination.
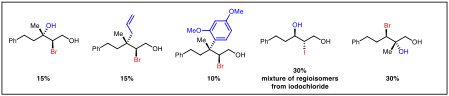
Figure 3. Products accessible from non-benzylic dihalides.
Conclusions and future work
To fully explore the synthetic utility of these newly discovered transformations, further optimization as well as a full investigation of the substrate scopes will be carried out. To introduce additional molecular complexity, studies into the displacement of the second halide will be conducted. As monohalides react orders of magnitudes faster than dihalides this task is predicted to be less challenging. With all these tools in hand, we will then have the opportunity to synthesize a wide range of biologically relevant compounds and motifs.
[1] (a) Hu, D. X.; Shibuya, G. M.; Burns, N. Z. J. Am. Chem Soc. 2013, 135, 12960–12963; (b) Hu, D. X.; Seidl, F. J.; Bucher, C.; Burns, N. Z. J. Am. Chem. Soc. 2015, 137, 3795–3798.










