Reports: UNI153776-UNI1: Development of a Palladium-Catalyzed Oxidative Aryl Transfer Reaction
 printer friendly
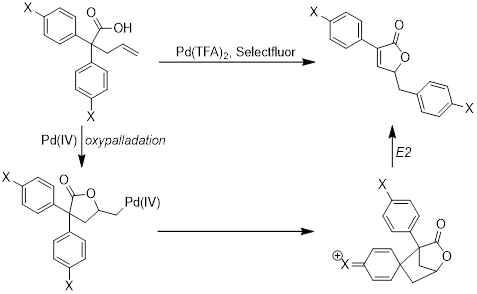
printer friendlyPalladium-catalyzed reactions have proven invaluable in the search for new and interesting reactions. The major area of chemical research in our lab has been in the development and exploration of reactions catalyzed by palladium in a high oxidation state. Our lab’s specific aim is the study of a novel reaction catalyzed by palladium(IV) that involves the transfer of an aryl group. This reaction is unprecedented under palladium-catalysis and provides efficient access to an interesting structural motif, while offering insight into the reactivity of high-oxidation state palladium. Our lab is working to explore the scope and mechanism of this reaction through careful examination of the steric and electronic requirements of the system. The information garnered from this project will be used to inform the development of additional new and novel palladium-catalyzed transformations.
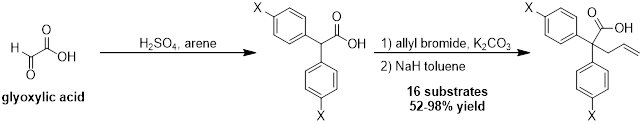
In previous years, we have optimized the reaction conditions and explored efficient methods to synthesize a variety of substrates. Substrate synthesis has been the limiting step of the process, as it requires three to four linear steps starting with the dehydration of glyoxylic acid to form diarylacetic acids. Direct allylation of the alpha-carbon is problematic for the more hindered substrates; therefore, we have adopted a two-step procedure that involves the allylation of the carboxyl group followed by a Claisen rearrangement. We have currently used this method to synthesize sixteen different substrates in moderate to excellent yields.
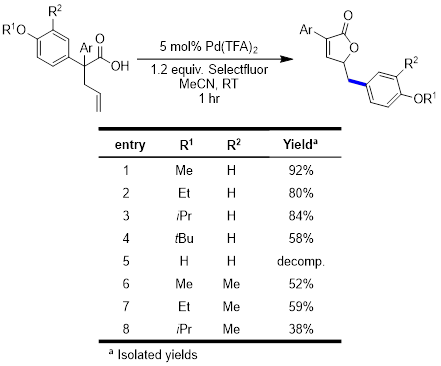
With an effective substrate synthesis and optimized conditions, our more recent focus has been on understanding the substrate scope with a special interest in garnering mechanistic information. Our current mechanistic hypothesis indicates that the formation of the α,β-unsaturated lactone product proceeds through a dearomatization process. In an attempt to study this proposed intermediate in greater detail, the reaction was run with various alkyl groups on the activating oxygen. Interesting, there was no significant impact on the course of the reaction for even labile groups such as t-butyl. In addition, only decomposition was observed when a phenol substrate was tested under the optimized reaction conditions. Current investigations are focused on further modulation of the substituents on the oxygen. In addition, we are currently investigating the stereochemical outcome of the reaction, with results forthcoming.
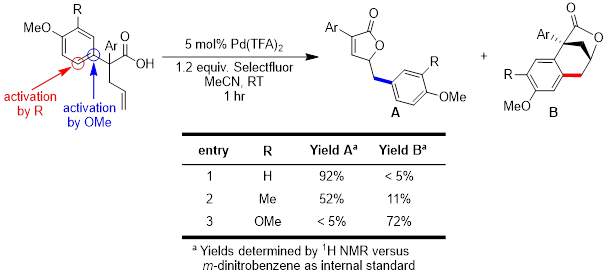
Initial studies have indicated that the position of the activating groups has a significant effect on the reaction outcome. Placement of an electron donating group in the meta-position leads to the formation of a tricyclic product instead of the desired rearranged product. Identification of the tricyclic product (B) is complicated by hindered rotation of exocyclic aromatic ring, confirmed by coalescence of 1H NMR signals at high temperature. Increasing the electron-donating capability of the R group on the aromatic ring significantly increases the formation of tricyclic product. The reaction is highly sensitive to groups on the ring, but the formation of the carbon-carbon bond is always favored at the most electron-rich site. We are continuing to explore this divergence in reactivity with less donating substituents, such as alkyl groups.
As we are located at a small liberal arts institution, all laboratory was conducted by undergraduate students. Generous ACS-PRF support has enabled three different students to have a fully funded summer research experience, while providing research supplies for an additional three. Two students who have worked on this research project have graduate studies in the chemical sciences, seeking both terminal Masters and Ph.D. degrees. Students have gained valuable laboratory experience, learning the technical skills as well as gaining a deeper understanding of chemistry.










