Reports: DNI656782-DNI6: Guiding Nucleation and Growth of Metal-Organic Frameworks
 printer friendly
printer friendlyMetal-organic frameworks (MOFs) consist of organic molecules that are connected by metal ions (MOFs) and form nanoporous crystal lattices. Because of their high porosity, these materials are outstanding potential platforms for gas storage, catalysis, separations, optoelectronic devices, and charge storage. However, specific applications make specific demands on a framework's pore size, crystal structure, stability, and chemistry. While synthesized MOFs display a large variety of these properties, no general guiding principles exist that allow the synthesis of a MOF with pre-programmed physical and chemical properties – in fact, the discovery of new MOFs usually proceeds through time consuming and oftentimes unproductive trial-and-error strategies. At the bottom of this challenge lies our limited knowledge about the fundamental microscopic processes that occur during the nucleation and growth of porous framework materials. In this project, we address this problem with a computer simulation study of porous framework formation. We use simple and efficient computational models of prototypical MOFs to identify the microscopic mechanisms underlying framework nucleation and growth, as well as their susceptibility to changes in experimental control parameters. The overarching goal of this project is to provide experimenters with firm guiding principles that enable the targeted synthesis of porous framework materials with desired structure, porosity and chemical composition.
Nucleation and growth of zeolitic imidazolate frameworks (ZIFs)
Which experimental parameters control the formation of different polymorphs? Do MOFs form through classical monomer addition or via a two-step mechanism involving amorphous precursors? We have chosen zeolitic-imidazolate frameworks (ZIFs), a prototypical family of MOFs, as our platform for answering these questions. ZIFs have a simple chemical composition (zinc ions and imidazole linkers, as illustrated in Figure 1a) and display many intriguing features of MOF formation, including pronounced polymorphism and evidence for non-classical formation pathways. Choosing the well-studied framework ZIF-8 as our starting point, we have developed a coarse-grained molecular model of ZIF-8 constituents, zinc ions and methyl-imidazolate linkers (Figure 1b). We have used this model to simulate spontaneous nucleation from supersaturated solution. Our simulations confirm recent in-situ SAXS results that suggested the possibility of a two-step nucleation mechanism involving an ensemble of amorphous clusters that grow and eventually crystallize (Figure 1c,f).
Our simulations furthermore predict the occurrence of a gyroidal polymorph with large, tunnel-like pores that has not yet been observed experimentally. While this new polymorph is metastable with respect to ZIF-8, it can be made stable by modifying the molecular shape of imidazolate linkers (Figure 1d-e). These results suggest molecular shape as a powerful tool in the design of novel MOFs. We are currently investigating chemically feasible ways of realizing the gyroid structure experimentally.
Biasing framework formation via solvent fluctuations
For ZIFs comprising unsubstituted imidazolate linkers, a large number of different framework topologies can be produced by small changes to synthetic conditions including temperature, monomer concentrations, and solvent composition. We hypothesize that solvent fluctuations can play a major role in determining framework structure. To investigate the role of solvent in the formation of ZIFs, we have developed a simple model that reproduces the relative enthalpies of different ZIFs measured in experiment, as well as amorphous and glassy phases of ZIFs. Together with solvent models of increasing complexity, we will perform free energy calculations to determine how nucleation rates of different ZIF polymorphs depend on the density, molecular size, and interactions of solvent molecules.
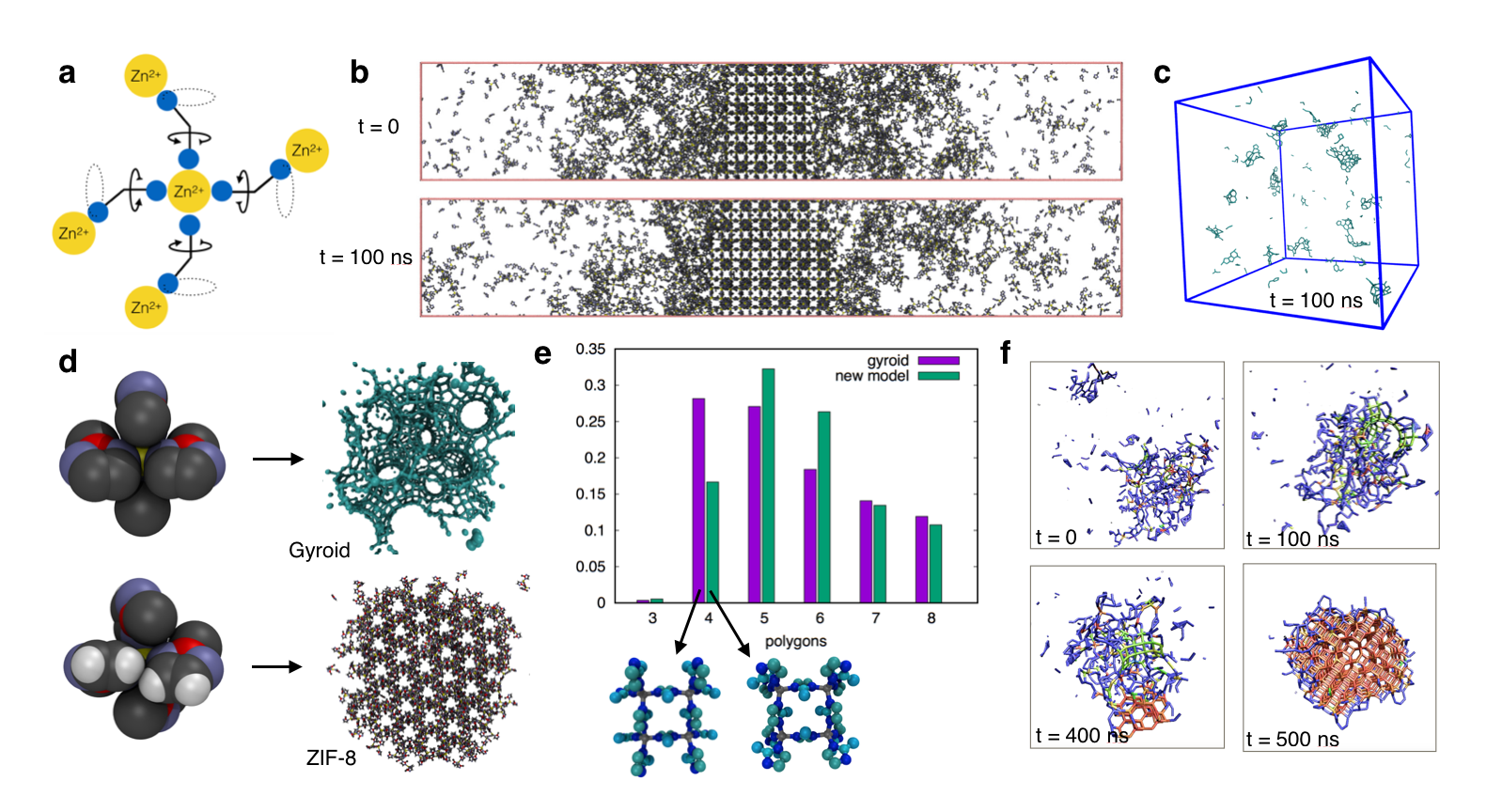
Figure 1: Modeling the formation of ZIF-8. (a) ZIF coordination environment. A central zinc ion is coordinated by four imidazolate linkers (shown only schematically as nitrogen atoms (blue) connected by line segments). For unsubstituted imidazolate linkers, the rotational freedom of linkers, indicated by arrows and dashed lines, gives rise to pronounced polymorphism of ZIF frameworks. For 2-methylimidazole linkers, ZIF-8 is one of only two known topologies, due to steric hindrance of linker rotations. (b) Snapshots from straightforward MD show the growth of ZIF-8 under large supersaturation. (c) Snapshot from MD under experimentally realistic supersaturation show the formation of amorphous clusters. (d) Framework formation is extraordinarily sensitive to details of molecular linker shape. A coarse-grained model treating hydrogen atoms implicitly invariably forms gyroid-like structures (upper panel). Explicit modeling of hydrogens (lower panel) disfavors gyroid-like molecular fragments energetically trough steric effects and results in spontaneous formation of ZIF-8. (e) Probability of finding different polyhedral motifs (triangles, squares, pentagons, etc.) in the early stages of framework formation, for the two models shown in (d). A large population of squares in simulations of the coarse-grained model is indicative of gyroid formation. (f) Snapshots from MD showing ZIF-8 formation. "Bonds" between nearest neighbor zincs are colored according to a tetrahedral order parameter: Green indicates gyroid-like fragments, orange indicates ZIF-8, and zincs with less than four nearest neighbors are colored blue.










