Reports: DNI755967-DNI7: Synthesis and Evaluation of Sequence-Defined Branched Polymers
 printer friendly
printer friendlyOur initial goal for this reporting period involved the assembly of branched sequence-defined macromolecules. Our next goal was then to use these constructs to investigate the effect of primary sequence and branching on macromolecular properties such as compactness and chain dynamics. We've since modified our approach to begin with an investigation into the compactness and chain dynamics of linear polymers, and use the knowledge derived from this study towards creating and studying branched systems. Herein, this report will detail our efforts into the development of physical methods to investigate the solution phase behavior of macromolecules.
Solution-phase characterization of macromolecular structure and dynamics can be challenging due to the small length- and time-scales, which can limit common experimental structural elucidation techniques. Herein, we have adapted and applied existing methods for the solution-phase characterization of sulfonated oligothioetheramides (oligoTEAs) as a function of oligomer length (2-12mer). We explored the effect of pendant group charge and backbone composition on the dynamics of the oligoTEA chain. Structural investigation was performed by variable temperature diffusion ordered nuclear magnetic spectroscopy (VT-DOSY), pulsed electron paramagnetic resonance (EPR), and molecular dynamics (MD) simulations. VT-DOSY most directly measures the macromolecular hydrodynamic radius. EPR and MD can both observe solution-phase dynamics with double electron-electron resonance (DEER) EPR quantifying the distance distribution between paramagnetic spin-labels. MD modeling elucidates how the oligoTEA chain evolves over time.
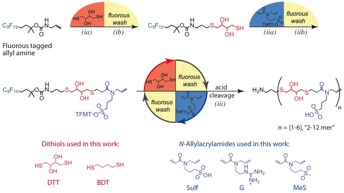
Figure 1. Oligothioetheramide assembly. TMFT - α-trifluoromethyltolyl protecting group.
All oligomers were HPLC purified after cleavage from the fluorous support and characterized via 1H NMR and LCMS. To explore the effect of each chemical group, additional oligomers were synthesized at the 10mer length with 1) a positively charged guanidine (G) group 2) a non-charged methyl sulfone (MeS) pendant group and 3) a methylene backbone using butanedithiol (BDT) (Figure 1).
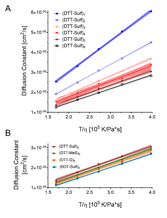
Figure 2. A. Translational diffusion coefficient versus viscosity normalized temperature of oligoTEAs. B. Diffusion coefficients of oligomers with different pendant and backbone groups as a function of temperature.
Characterization of oligoTEAs by VT-DOSY visualizes macromolecular diffusion as a linear function of viscosity normalized temperature (Figure 2). This linearity rules out any intramolecular transitions as a function of temperature and suggests minimal intermolecular interactions (e.g., aggregation, repulsion). Diffusion is affected by the hydrodynamic size, shape, and hydration state of a given macromolecule. The data in Figure 2 follow expected trends in that these macromolecules diffuse quicker at higher temperatures or shorter synthetic length. The oligomers with hydroxylated (DTT) backbone diffused slightly faster than the aliphatic backbone (BDT). This result can be rationalized by a number of factors including differences in shape, hydration effect caused by the more hydrophobic BDT backbone, or stronger hydrogen bonding in the DTT hydroxylated backbone resulting in a more compact faster diffusing oligomer.
To observe the dynamics of the synthesized oligoTEAs in an aqueous environment, DEER EPR distance measurements and molecular dynamics (MD) simulations and were completed. DEER measurements were performed on dispin-labeled oligomers. Single oligomer chains were simulated for 50ns in a water-filled box with periodic boundary conditions in an NVT ensemble to represent a dilute oligomer solution. Specifically, the (DTT-Sulf), (DTT-MeS), and (BDT-Sulf) oligoTEAs were simulated at all lengths (2-12mer) to aid in the visualization of trends as a function of synthetic length. To validate the simulations, the translational diffusion constant was calculated and compared to VT-DOSY measurements, revealing good agreement.
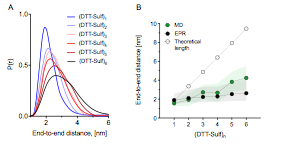
Figure 3. A. Distance reconstructions of the end-to-end distance distribution function generated from DEER EPR of the (DTT-Sulf)1-6. B. Mean end-to-end distance versus synthetic length from DEER distance reconstruction and MD simulation. Transparent bands represent the full-width at half maximum (FWHM) from EPR or standard deviation from the MD simulation time.
With respect to the (DTT-Sulf)1-6 series (Figure 3A), the probability distribution from EPR data shows an increase in end-to-end distance with increasing oligomer length, as well as a concomitant increase in conformational freedom as measured by the full width of half max (FWHM). The mean end-to-end distance of the oligomers calculated by MD is also in reasonable agreement with the EPR data, showing a modest increase with oligomer size. A slight overestimation of the MD is observed with the (DTT-Sulf)5,6, but is within the experimentally observed conformational ensemble (Figure 3B). Overall, this data highlights oligomer flexibility, especially when considering their fully extended theoretical length showing on average ~50% collapse (Figure 3B).
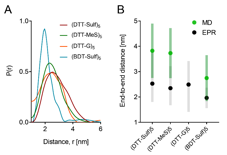
Figure 4. A. Distance reconstructions of the end-to-end distance distribution function generated from DEER EPR. B. Mean end-to-end distance versus molecular weight from DEER distance reconstruction and molecular dynamics simulation. Error bars represent the full-width at half maximum (EPR) or standard deviation (MD).
End-to-end distance measurements from DEER and MD were used to visualize the individual contributions of backbone hydroxylation and pendant groups, revealing a larger effect from the backbone. 10mers with a methylene backbone, i.e., (BDT-Sulf)5, positive charge (DTT-G) 5, and neutral charge (DTT-MeS) 5 were characterized (Figure 4). The EPR and MD results both show that the pendant groups have minimal influence on the average end-to-end distance and the conformational flexibility of the 10mer oligoTEAs. The methylene backbone of (BDT-Sulf)5 results in a smaller end-to-end distance in both DEER and MD experiments. This effect could be rationalized by greater hydrophobic collapse without backbone hydroxylation. However, VT-DOSY suggests that the (BDT-Sulf)5 diffuses slower than all other 10-mers with pendant group modifications (Figure 2). Together these results can be rationalized by two possible explanations: 1) the hydroxylated backbone participates in intramolecular interactions rather than hydration as would be expected, aiding collapse by attractive forces (i.e., hydrogen bonding) to result in a smaller hydrodynamic size than the BDT-Sulf, and/or 2) the spin-probes are participating in hydrophobic collapse and thus locate closer to one another in BDT-Sulf. Though possible, the latter is unlikely since the MD simulations performed on oligoTEAs predict a similar trend without the spin probes present on the molecular structure (Figure 4B).
While low resolution, these techniques outlined in this report have provided insight into the ensemble of highly flexible oligoTEAs. As such, this methodology can be applied to the structural characterization and development of flexible and branched macromolecules.










