Reports: ND456519-ND4: Variable Temperature NMR Study of Proton-Bound Dimers. Role of Dispersion in Solvatophobic Effects on Bond Strengths
 printer friendly
printer friendlyThe PRF-funded project has produced informative results, which, when combined with gas-phase and computational work on the same systems, gives a comprehensive assessment of the role of dispersion in the strength of the chemical bond. A first full paper has just appeared, detailing the first conclusions.
“Attenuation of London Dispersion in Dichloromethane Solutions,” R. Pollice, M. Bot, I.J. Kobylianskii, I. Shenderovich, P. Chen, J. Am. Chem. Soc. 2017, 139(37), 13126–13140. DOI:10.1021/jacs.7b06997.
The work is a direct output of the ACS-PRF funding. In comparison to the proposal, we investigated a much larger set of proton-bound dimers of pyridines, tertiary amines, and quinolines, 36 in all, and, in addition to the proposed variable temperature NMR work, we executed isothermal calorimetric measurements to ensure that the measured equilibrium constants were correct. The conclusion of the paper, is reproduced here:
In this work we report an extensive study of bond dissociation equilibria of a wide variety of proton-bound dimers in the gas-phase and in solution by T-CID measurements, computational methods and NMR studies. We conclude that London dispersion becomes very significant for medium-to-large molecules in the gas-phase, which was observed directly by experiment, and is always accompanied by significant contribution of repulsive Pauli exchange, which, however, does not fully compensate the attractive contribution of dispersion. The significant attractive contribution of dispersion, despite being attenuated by about 70%, still transfers into dichloromethane solution temperatures that are relevant for chemical reactions, at least for the chosen test systems, showing that altering London dispersion can indeed be a useful design principle to tune molecular stability for chemical processes in solution. Furthermore, currently employed implicit solvent models are shown to describe attenuation of dispersion in solution inadequately, especially when the corresponding contribution becomes large, showing that alternative theoretical approaches and models are required in that regard for a proper estimation of energies. While we have chosen a specific test system for this study, our results and conclusions are not specific to that type of system. The contribution of dispersion to the gas-phase bond dissociation energies in molecular complexes decorated with increasingly large interacting hydrocarbon substituents is expected to become large for any molecular complex. Additionally, the observation that intermolecular London dispersion, while largely attenuated, still transfers into a polar organic solvent like CH2Cl2 is likely more general, as well, and we expect a very similar trend for other comparable organic solvents. Furthermore, the spectacular failure of implicit solvent models to predict the Gibbs free energy of dissociation in solution, suggests that these models are inadequate to describe dispersive solute-solvent interactions. Again, this is expected to be applicable not only to our test system, but in general to molecular complexes having sufficiently large interacting hydrocarbon substituents. Considering these points, and the wide variety of systems for which a DFT+PCM or a DFT+COSMO computational approach has been applied to in literature, we claim that our conclusions are widely applicable to homogeneous catalysis and supramolecular chemistry in polar organic solvents. Further work continuing gas-phase measurements using T-CID, investigating proton-bound dimer equilibria in different solvents and exploring alternative theoretical approaches to estimate Gibbs free energies of solvation are underway in our group.
It summarizes well our findings that attractive dispersive interactions in the gas phase are largely quenched by solvation effects in dichloromethane solution. This is a significant, general, and potentially controversial finding. We are continuing the project, as proposed, with a parallel set of measurements in fluorous solvents, whose weak intermolecular dispersive interactions should less completely cancel out the hydrocarbon-hydrocarbon dispersive interactions seen in the gas phase. Accordingly, we expect much stronger binding for the dimers in fluorous solution. This will be the “smoking gun” experiment! The fluorine lock budgeted in the PRF proposal has been acquired and installed. Presently, we are synthesizing a highly fluorous BArF anion, depicted below,
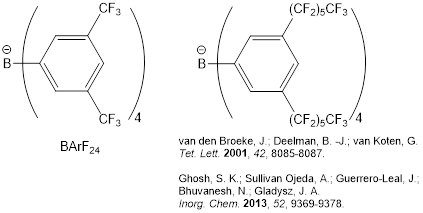
which has been cited in the literature as conferring complete solubility to simple alkali and alkaline earth cations in perfluoromethylcyclohexane. Given that solubility (actually the lack thereof) of the proton-bound pyridine dimer cations in fluorous solvents is the principal technical barrier to our further measurements, and the counterion in published work is the so-called BArF24 anion, we expect that the highly fluorous anion will enable the next phase of the project in which we measure monomer-dimer equilibria for the same series of 36 proton-bound dimers as we did in dichloromethane. The synthesis of the highly fluorous anion has been achieved on millimole scale. The present task is to get it pure enough for the demanding experimental studies.










