Reports: DNI356083-DNI3: Mononuclear Iron Mercaptoimidazolyl Complexes for the Reductive Coupling of Isonitriles
 printer friendly
printer friendlyThe long-term goal of our work in this area is to understand how enzymes achieve complex multi-proton and –electron transfers under mild conditions. The ability to control such steps has implications for the design of catalysts relevant to energy storage and fuel production. While our long-term goals targets the development of homogeneous catalyst systems for the reductive coupling of CO, our initial studies in this area have used isonitriles (CNR) as models for CO because they provide additional spectroscopic handles while eliminating the need for handling gases and pressurized reactors in initial studies.
Prior Work. Our preliminary work in this area (completed before this award was received) focused on the synthesis and reactivity of a series of thione-ligated isocyanide-bound iron complexes, [TmRFe(CNR')3](OTf) (R = Me, Ph, mesityl; R' = tBu, Bn, Cy). These complexes are designed to structurally mimic nitrogenase while enabling systematic modification of key features to evaluate their role in CNR reductive coupling. We have found that the reaction of [TmMeFe(CNtBu)3](OTf) with KC8 as the reductant and H2O as a proton source generates 1,3-di-tert-butyl urea, a new isocyanide derived coupling product (Scheme 1). A series of mechanistic experiments, including amine crossover studies and 18O2 labeling experiments, suggest a pathway involving initial reduction of CNtBu to tBuNH2, a key intermediate. A series of labeling experiments indicate air to be the source of the urea oxygen while the urea carbon is derived from the isocyanide. Crossover experiments indicate a key role for H2NR in the formation of the di-substituted urea and we proposed a pathway for urea formation that involves a diaminocarbene intermediate that is formed from H2NR attack on a bound isocyanide ligand. This work was submitted to publication prior to our PRF funding and lays the groundwork for our current and ongoing studies described below.
|
Scheme 1. Redox-coupling reactivity of [TmMeFe(CNtBu)](OTf). |
.](abimages/Paper_15033_abstract_37438_0.png)
Reactive Intermediates. Much of our work during the current year has been focused on improving our understanding of the nature of the reduced species and the possible role of the R and R' groups in influencing the conversion of CNR to di-substituted urea. During the reaction of [TmRFe(CNR')3](OTf) to form di-substituted urea a color change from orange to yellow is observed. This change was associated with the reduction of the iron ion towards an iron(I) species. To further probe the nature of the iron species present, [TmMeFe(CNtBu)3](OTf) (1) was reduced with KC8 in the absence of water and oxygen. Under these conditions, the same color change from orange to yellow was observed and the conversion of 1 was measure using UV-Visible spectroscopy (Figure 1).
|
Figure 1. Absorption spectra of the reaction 1 with KC8 in THF over 23 h. |
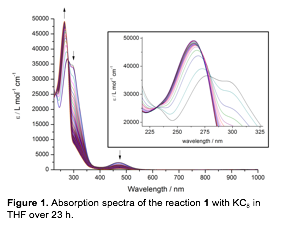
Eletrochemical measurements have been conducted on a small series of the [TmRFe(CNR')3]+ complexes (R = Me, Ph, Bn and R' = tBu, Bn, Cy, Ad,) and reveal that the spectroscopic changes likely arise from two isocyanide based reductions followed by a single reduction of the iron center to iron(I). The potentials were determined by square wave voltammetry and it appears that the potentials vary only slightly based on the substitution of the Tm ligand, while they are unaffected by the isocyanide. Our data combined suggest that during prior to urea formation, the isocyanides undergo two subsequent reduction events followed by the reduction of the iron center to an iron(I) species. The latter occurs at a potential low enough that the reaction requires the strong reductant KC8, while Cp2Co is unable to effect this reductive coupling reaction.
Synthesis and Reactvity of [TmFe(CO)3](OTf). The reduction of CNR to RNH2 suggests that these TmFe complexes may also facilitate the challenging cleavage of the C-O bond of CO. We are now well poised to explore the reactivity with the corresponding CO complexes which we have isolated in 59% yield from the reaction of Fe(OTf)2 with NaTmMe under 90 psi of CO (Scheme 2). We are currently exploring the reactivity of these carbonyl complexes in the presence of a strong reductant and a proton source.
|
Scheme 2. Synthesis of [TmRFe(CO)3](OTf) complexes. |
 complexes.](abimages/Paper_15033_abstract_37440_0.png)
We anticipate that this work will allow for a fundamental understanding of the catalyst requirements for CO reduction and coupling and will lay the foundation for a detailed mechanistic understanding of the multiple protonation and reduction events accomplished in the enzymatic reduction of CO and other small molecules.










