Reports: DNI656806-DNI6: Computational Study of Fundamental Organic Transformations Catalyzed by Metal Oxides
 printer friendly
printer friendlyOVERVIEW
Exploration of metal oxide mediated reactions is critical to expanding the development of new approaches for organic transformations, which are essential in the petroleum industry. The objective of this project is to explore fundamental aspects of structure in metal oxide chemistry and the catalytic function such systems serve in fundamental organic transformations.
During the first year of the funding period, the PI’s team (a) completed benchmark studies supporting the importance of locating minimum energy structures using spin projection models when calculating spin-state energy differences; (b) demonstrated a dependence of the potential energy surface’s curvature on spin contamination; and (c) began studying the structure and bonding of lanthanide hydroxides.
CURRENT AND ONGOING WORK
The objectives for the first year of this computational project were to validate spin projection models for use in metal oxide chemistries, understand the extent to which spin contamination might degrade calculated potential energy surfaces, and to begin examining structure and bonding of lanthanide oxides.
Benchmarking Spin Projection Models for Computing Spin State Splitting
Many of the systems of longer-term interest will present significant signs of so-called spin contamination when treated with common electronic structure models such as approximate density function theory (DFT). While the use of spin projection methods for such studies is not necessarily uncommon, multiple researchers have reported worse using spin projection as comparted to not. Interestingly, it does not appear that geometry optimization using spin projection corrected energy surfaces have been carried out as part of prior studies. With this in mind, and using analytic gradient codes recently developed in their lab, the PI’s team explored the impact of spin projection on geometry optimization and its resulting effect on spin state splitting energies.
Two validation tests have been carried out as part of this study. The first set is comprised of eight binuclear metal complexes with experimentally characterized exchange coupling constants. As shown in Fig. 1, optimization using the Approximate Projection (AP) model during geometry optimization provides very good agreement with experiment using most of the tested density functionals. This strongly suggests that spin projection should be used in the study of spin-coupled complexes. Importantly, multiple metal oxide clusters the PI’s group plans to study in the future belong to such a class.
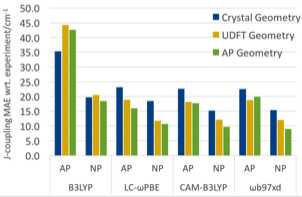
Figure 1. Mean absolute errors (MAEs) of J-coupling constants calculated using different schemes for optimization and for calculating J. Four different density functionals have been included in the study.
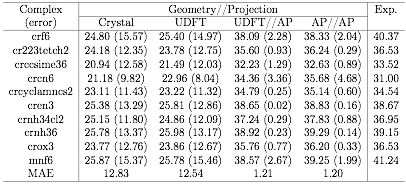
As a second validation test, a set of spin-crossover cases was considered. Focusing on octahedral species with d3 occupation, which one would expect the AP model should work, Table 1 shows that spin projection does provide excellent agreement with experiment. Using crystal structure geometries for the calculations or optimizing geometries on spin contaminated potential energy surfaces yields average spin-crossover energy errors > 10 kcal/mol. On the other hand, using spin projection reduces average errors to roughly 1 kcal/mol.
Table 1. Spin cross-over energies for a test set of d3 transition metal species. Values in parentheses give errors relative to experimental results. All energies are given in kcal/mol.
The results of this first validation study strongly support the computational protocol planned for the remainder of the grant project.
Data Analytic Examination of Spin Contamination Effects on Vibrational Frequencies
The second benchmark study specifically explores the effect of spin contamination on calculated vibrational frequencies of small transition metal systems. This study has been motivated by recent observations during the PI’s group’s applications studies. Specifically, it has been empirically found that spin projection often has a strong effect on force constants, even in cases where spin projection seems to have little effect on energy and/or geometry.
A data-analytics approach has been employed to explore factor(s) -- functional selection, basis set choice, elements involved, degree of spin contamination, etc. -- most significantly affect agreement between calculated and experimental vibrational frequencies. The current test set includes a mix of metals and molecules exhibiting varying degrees of spin contamination. Additionally, the set includes a mix of ground spin states -- roughly a third have singlet ground states and with others having doublet through septet ground states. With this test set, benchmark calculations have been carried out using a variety of model chemistries. The results show that spin contamination is the only considered independent variable showing strong correlation with errors in calculated vibrational frequencies. A complete and full analysis of this data set is near completion and should be published soon. In the future, the study will be expanded by including a few bimetallic molecules and will also expand the range of models considered (including additional functionals, post-SCF, much larger basis sets, and effective core potentials).
Initial Studies of LnOH
Motivated by new work by experimental collaborators, the PI’s group has begun exploring the structure and bonding of lanthanide hydroxides. This family of compounds will provide initial insight to the structure and bonding of these types of bonds and will prepare the PI’s group for investigations of lanthanide oxides, including reactivity of these systems with small organics.
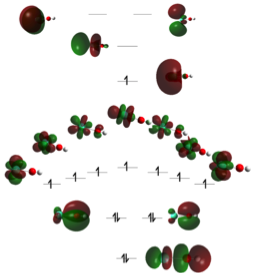
Figure 2. Molecular orbital diagram for EuOH, which serves as an example system for all LnOH compounds.
There are two important results from this study. First, the geometries of these triatomics are linear rather than bent. This trend holds across the full lanthanide series, which is in contrast to transition metal hydroxides where the bond becomes increasingly bent as one moves across the series. Second, bonding analysis (see, for instance, the molecular orbital diagram in Fig. 2) indicates the LnOH exhibit high degrees of covalency and the Ln-OH bond is best described as a triple bond. It is anticipated that these new bonding observations will be critical in the unique structure and reactivity often observed in lanthanide oxide surfaces and transition metal oxides doped with lanthanides. Consideration of such questions will be addressed more specifically in the next year.
Building from this work, the PI’s group will explore LnO clusters as well as metal oxide clusters that include both transition metals and lanthanides during the next year of funding.










