Reports: DNI455595-DNI4: Unraveling Heterocycle-Promoted Hydride Transfer Mechanisms for Energetically Efficient Fuel and Petrochemical Production
 printer friendly
printer friendlyAbstract:
Our society's future hinges on scientists and engineers being able to develop sustainable technologies. To this end we are researching how to sustainably generate renewable fuels and chemicals from CO2. Aromatic N-heterocycle (ANH) molecules have been implicated in energetically efficient conversion of CO2 into molecules such as CO, formate, and methanol, but the exact roles of these heterocycles remain unknown. Recent experimental reports of these systems (e.g. by SavŽant and co-workers) have raised concerns that ANH molecules may not actually have any role, but we have studied these systems to learn what environmental conditions would allow energetically efficient hydrogenations of CO2 and how one should best computationally model these hydrogenations. We use quantum chemistry to predict thermodynamic energies of hypothetical reaction intermediates to identify values of pH and applied potential where those intermediates have similar free energies. When this happens, the hypothetical reaction steps would be energetically efficient in accordance with the Sabatier principle. Most of our thermodynamic analyses have been discussed in our report from last year as submitted work, and those papers have now been published. We now summarize our results from the second stage of the project focusing on how to model kinetics of these processes. From this work, we aim to provide guidelines to advance the engineering of renewable fuel catalysts.
Progress:
Our objectives have somewhat changed since the start of this project. The proposal originally planned to study ANH hydride transfer mechanisms to reduce CO2, but it became apparent from studies in the literature these molecules might not actually be catalyzing CO2 reduction. Furthermore, the level of modeling we initially planned was found to not be reliable for modeling barriers for entire reaction mechanisms, and thus would not be appropriate for obtaining kinetic parameters for microkinetic modeling studies.
We investigated the energetics of hydride transfers to CO2 in aqueous solutions using atomistic modeling with explicit solvation modeling at an ab initio level. NaBH4 was used instead of a dihydropyridine molecule because it is a very strong hydride donor, and it also consisted of only five atoms which made computational modeling easier. The barriers we found using that approach were qualitatively different (Figure 1) than barriers identified using more conventional modeling using continuum solvation modeling (Figure 2). Instead of a single barrier being 0.7 eV high that was found using simple modeling with continuum solvation, barriers with explicit solvation were significantly lower, 0.43 eV, while a metastable BH3-H-CO2 intermediate was also found with explicit solvent modeling. We attribute the differences to continuum solvation modeling alone not adequately local solvation effects that stabilize reaction intermediates and allow lower energy pathways with concomitant proton transfer reactions involving nearby solvent molecules.
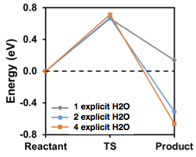
Figure 1. Hydride transfer reaction pathway modeled using a minimal number of explicit solvent molecules and continuum solvation models.
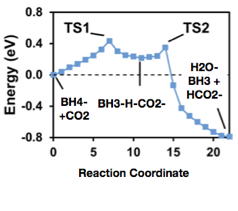
Figure 2. Hydride transfer reaction pathway modeled using explicit solvation models.
We originally suspected hydride transfers would pose a challenge for standard Kohn-Sham density functional theory since the latter is known to unphysically model charge transfer states and higher levels of theory (or embedding calculations) would be needed. We learned this was not true, and different levels of quantum chemistry theory all resulted in very similar reaction energetics within about 0.1 eV. The most significant requirement for modeling hydride transfer barriers was whether explicit solvation treatments for the first coordination shell of the reacting molecules was used (including the Na+ counter ion). Indeed, using a continuum solvation model in place of all solvent molecules outside of the first solvation shell resulted in close agreement with fully explicit calculations. This opens a clearer path forward for how to model hydride transfers in electrolytes. This work was published in J. Phys. Chem. B in late 2016.
We also investigated the role of free energy corrections on these pathways, modeled more rigorously (but much more computationally intensively) using potentials of mean force. Importantly, we find that explicitly modeling free energies due to solvent configurations and thermal effects at 298 K result in significantly different energy profiles compared to energy profiles modeled at 0 K. The large differences may be because 0 K pathways obtained from nudged-elastic band (NEB) calculations were modeled using a metastable solvent configuration, but by accounting for solvent configurations lower energy pathways could be identified. Interestingly, the reaction coordinates of the reaction pathways themselves modeled at 0 and 298 K were quite similar. This shows mean force simulations are needed. While NEB energetics may not be reliable, potentials of mean force can be automated for pathways discovered using NEB calculations. This work has been accepted in ChemPhysChem in a special issue on CO2 utilization.
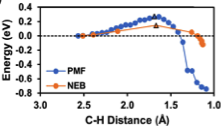
Figure 3. Comparison of reaction energies obtained using NEB calculations at 0 K and potentials of mean force at 298 K.
In other efforts, we validated that Pourbaix diagrams can also identify energetically efficient catalysis on a system not involving ANH molecules. We studied CO2 reduction on tin electrodes whose surfaces are known to form tin oxide upon exposure to air, but it is unclear if that tin oxide is then reduced over the course of CO2 reduction. We modeled this system and then predicted alloying dopants that could be added to tin to further reduce overpotentials for CO2 reduction. This work was published in J. Mater. Chem. A as a contribution to that journal's "Emerging Investigator" issue in 2017.
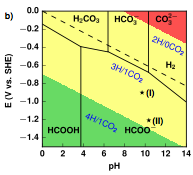
Figure 4. Overlaid Pourbaix diagrams for CO2 reduction on SnO2 surfaces. Stars denote experimental conditions where CO2 reduction occurs most efficiently.
Future Plans:
Now that we have identified that potentials of mean force are required for reliably modeling hydride transfers in solution, we are now investigating how to accelerate these simulations so that ab initio level treatments can be used to study potential-dependent reaction mechanisms for CO2 hydrogenations. In January 2017, this topic was awarded funding as part of the PI's NSF-CAREER project over the next five years. A central component of that project will be understanding how to accelerate calculations needed for reliable modeling of reaction mechanisms for CO2 hydrogenations.










