Reports: UR455293-UR4: Shedding Light on Diradical Forming Cyclizations and Cycloaromatizations
 printer friendly
printer friendlyGoals: In the second year of this grant two areas related to diradical-forming cyclization reactions were examined. First, substituent effects on photo-Bergman cyclization of 5,6-diethynylquinoxaline derivatives were examined in effort to optimize photocyclization yields of our designed quinoxalenediynes. Second, novel enyne-enones were prepared to examine thermal reactivity to determine if diradical cyclization channels similar to enediynes and enyne-allenes are available to these poly-unsaturated systems.
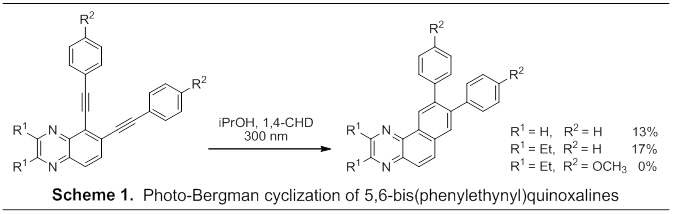
Results: We previously reported the successful photo-Bergman cyclization of 5,6-bis(phenylethynyl)quinoxaline to 8,9-diphenylbenzo[f]quinoxaline in 13% yield, a modest improvement in yield compared to 1,2-bis(phenylethynyl)benzene. In effort to further improve yields of photo-Bergman cyclization we examined modifications to both the quinoxaline core as well as the phenylethynyl substituent (Scheme 1). First, incorporation of ethyl substituents at the 2 and 3 positions of the quinoxaline scaffold leads to a 30% improvement in reaction yield. It should be noted, however, that the improved yield was primarily the result of increased recovery of unreacted quinoxalenediyne starting material as the isolated mass of photo adduct from the parent and diethyl substituted quinoxalenediynes were comparable under identical reaction conditions. We attribute more rapid consumption of the parent 5,6-bis(phenylethynyl)quinoxaline to Minisci reactions adjacent to the ring nitrogen atoms as higher molecular weight alkylated adducts were observed by GCMS analysis. As a second modification, para-methoxyphenyl substituents, shown to improve photo-Bergman cyclization yields of 1,2-bis((4-methoxyphenyl)ethynyl)benzene, were introduced. Interestingly, the addition of 5,6-bis(methoxyphenyl) substituents led to no observed photo-Bergman cyclization as the electron rich quinoxalenediynes rapidly degraded upon photolysis. A similar result was observed upon replacement of the 2,3-diethyl substituents with a fused phenanthrene ring. The exact nature of how these electron donating groups inhibit photocyclization is not known at the present time. 1H NMR data indicates slow rotation of the methoxyphenyl substituent suggesting extended conjugation to the out-of-plane pi-system which, in turn, reduces conjugation of the 4-methoxyphenyl substituent to the in-plane pi-system of the enediyne. As a result, irradiation at 300 nm excites the out-of-plane pi-system as opposed to the enediyne in-plane pi-system required to facilitate photo-Bergman cyclization. We have recently completed syntheses of the related naphthalenediynes and are currently examining photochemical reactivity to further probe this effect.
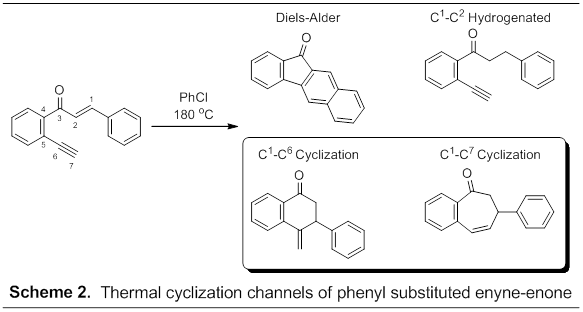
In a second approach to study diradical forming cyclizations we synthesized and examined thermal reactivity of the phenyl-substituted enyne-enone 2'-ethynylchalcone (Scheme 2). Thermolysis of this enyne-enone in chlorobenzene at 180 oC readily affords a tandem intramolecular dehydro-Diels-Alder cycloaddition followed by rapid dehydrogenation to afford the benzo[b]fluorenone adduct. Upon addition of 1,4-cyclohexadiene, however, additional products are formed including the C1-C2 hydrogenated product, C1-C7 diradical cyclization product, and a fourth product speculated to be the corresponding C1-C6 cyclization product. Unfortunately, the product mixture has proven difficult to separate and, as a result, we have only isolated and fully characterized the Diels-Alder and C1-C7 thermal cyclization products. Efforts are ongoing to purify and characterize the C1-C6 cyclization product. Finally, no evidence of the corresponding C2-C7 or C2-C6 cyclization products has been observed.
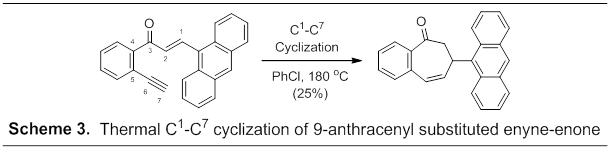
In effort to prevent the undesired Diels-Alder/dehydrogenation reaction pathway and simplify the product mixture we examined thermolysis of the related anthracen-9-yl derivative (Scheme 3). With Diels-Alder cycloaddition effectively blocked, the C1-C7 product was isolated as the major product in 25% yield. No evidence of alternative diradical cyclizations nor Diels-Alder cycloaddition products were observed. A similar strategy to limit the Diels-Alder/dehydrogenation pathway with a 2,4,6-trimethoxyphenyl substituent located at C1 has not afforded diradical cyclization products upon thermolysis to date.
Isolation of C1-C7 adducts from these enyne-enone derivatives confirms our calculations and represents a previously unreported diradical cyclization pathway similar to their enediyne and enyne-allene cousins. We are currently exploring the scope of this new reaction channel with alternative C1 and C7 substituents along with modified reaction conditions (temperature, 1,4-cyclohexadiene equivalents, solvent, microwave versus traditional heating). In addition, year three of the project will examine the synthesis and thermal reactivity of related enyne-ynone derivatives.
Impact on Undergraduates: During the 2016-2017 academic year a total of ten undergraduate chemistry majors worked on this project with four undergraduate students receiving their degree in spring 2017. In addition, two undergraduate students worked in the Gherman research group in our department conducting computational studies on enediyne and enyne-enone derivatives we prepared in the laboratory. Combined, their work resulted in six presentations at the spring 2017 National ACS meeting. In addition, three undergraduate students who worked on this project during 2016-2017 and two undergraduate students who worked during 2015-2016 are listed as coauthors on a full manuscript submitted to The Journal of Organic Chemistry (submitted September 2017). Finally, the first undergraduate author listed on the submitted JOC paper participated in our campus McNair’s Scholar Program designed to help prepare underrepresented students for admission to and completion of a Ph.D. program. This student graduated in spring 2017 and is currently enrolled in the Ph.D. program at UT Austin.
Impact on PI's Career: Conducting high quality research with undergraduate students is one of the key missions of our department. Funding from the ACS-PRF grant has allowed me to continue a strong tradition of undergraduate research with twelve undergraduates participating in this research to date. In addition, a number of small campus grants have been funded to support this research including faculty release time and undergraduate summer research stipends to supplement ACS-PRF funding. The PI has also been nominated to receive a Research and Scholarly Activity Award on campus for research and publications with undergraduate students. This award has also extended my collaboration with Benjamin Gherman (computational chemist in our department) and has sparked new collaborations with a biochemist in the department (Mary McCarthy-Hintz) to examine DNA binding and cleavage studies as part of a campus NIH RISE grant.










