Reports: DNI154824-DNI1: Site- and Enantioselective Electrophilic Halogen-Transfer Processes
 printer friendly
printer friendlyTwo powerful approaches promise to transform strategic small molecule assembly: late-stage atom-transfer processes and cross-coupling reactions. Both of these types of transformations enable previously unfeasible retrosynthetic disconnections, and this grant has supported advances related to both of these technologies. We have specifically targeted the development of new pre-catalysts to enable previously infeasible fluorination reactions. To this end, assays have been developed to study fluorine installation into sp3-hybridized carbon centers and the competitive oxygen-atom transfer process. With these assays in hand, we have interrogated previously reported fluorination methods to investigate the efficiency of fluorine transfer with a slightly broader range of metal-ligand complexes. Additionally, in the course of these efforts, we have developed selective, serial and exhaustive Suzuki-Miyaura reactions of polychloropyridines with alkylpinacol boronic esters. Consequently, this grant has laid the foundation to enable currently unfeasible fluorination processes, and has streamlined important and previously challenging cross-coupling reactions.
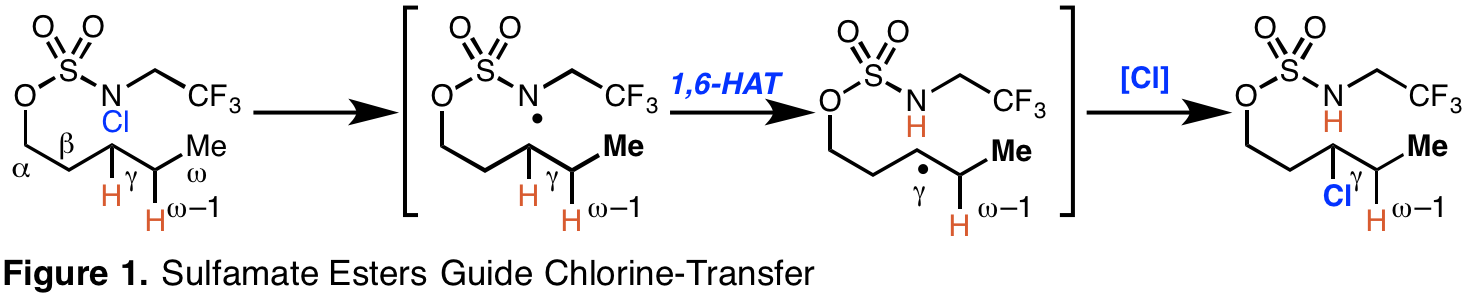
Sulfamate Esters Guide Chlorine-Transfer Reactions. Masked alcohols are ideal as candidates to be directing groups given the high frequency of hydroxyl groups in readily available fine chemicals. Nevertheless, there are no general, efficient approaches that employ a masked alcohol to guide aliphatic gamma-C-H functionalization.We have identified a novel strategy to alter the traditional position-selectivity of hydrogen-atom transfer processes. Specifically, we have established that pre-functionalized sulfamate esters direct site-selective aliphatic gamma-C-H atom-transfer in the course of a Hoffman-Loffler-Freytag-type chlorination reaction (Figure 1). This guided alkane C-H chlorination reaction proceeds through a chain-propagation mechanism.
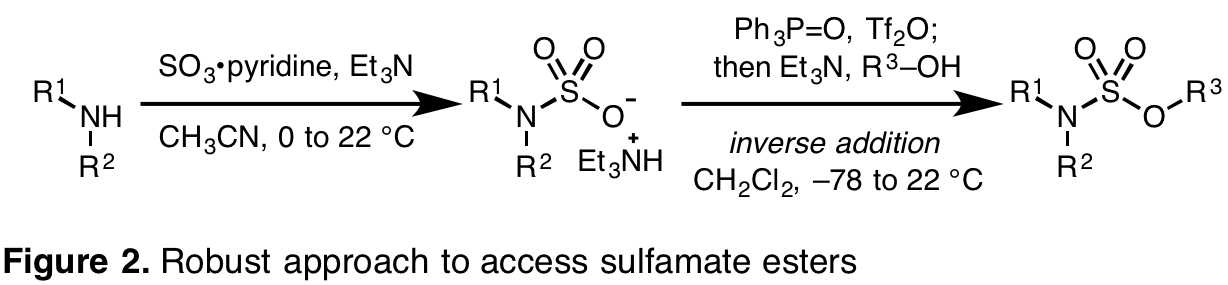
N-Substituted Sulfamate Esters Can Be Generated From Sulfamic Acid Salts Upon Activation with Triphenylphosphine Ditriflate. To enable investigations into sulfamate ester-guided chlorine-transfer reactions, we have developed a practical strategy to generate differentially N-substituted sulfamate esters. We predicted that initial amine sulfamation with a sulfur trioxide complex would furnish a sulfamic acid salt. Use of sulfamic acid salts has allowed us to investigate an array of esterification strategies to install the S-O bond of the sulfamate ester. Based on this approach, triphenylphosphonium ditriflate has been identified as an appropriate activating agent to enable trapping with a range of nucleophiles. This protocol can be employed to furnish differentially substituted sulfamides (Figure 2). Ultimately, these investigations will be extended to maximize their impact on position-selective alkane C-H functionalization technologies.
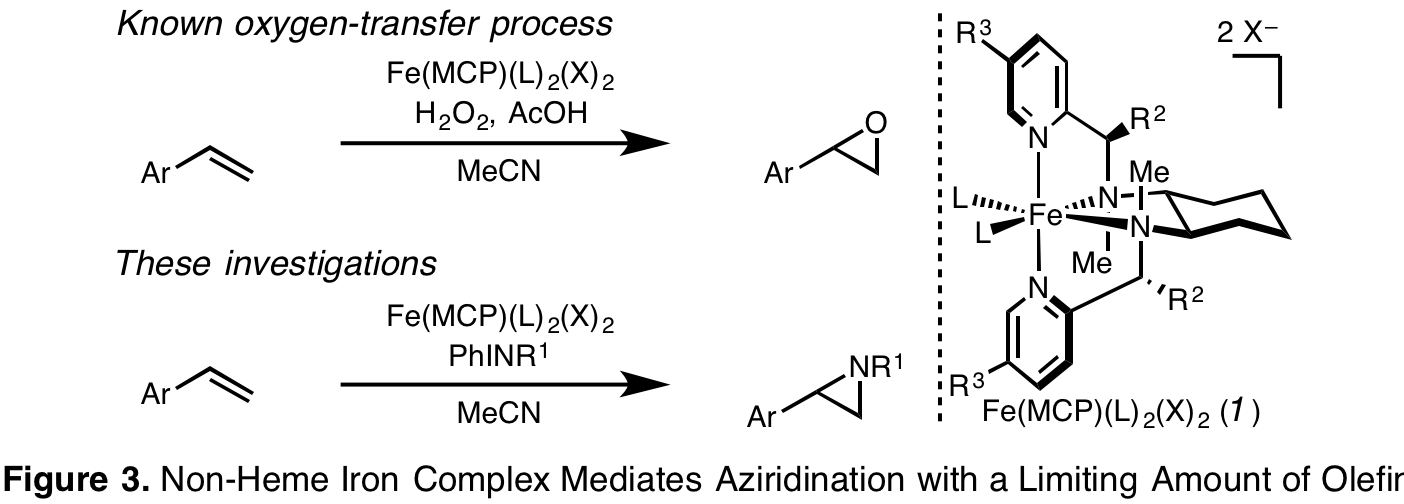
Molecular Non-Heme Iron Complex Mediates Aziridination With a Limiting Amount of Substrate. Molecular non-heme iron complexes catalyze remarkable processes, converting alkenes by dihydroxylation or epoxidation and aliphatic hydrocarbons by hydroxylation or dehydrogenation into higher value compounds. Fewer technologies advance nitrogen-transfer processes mediated by non-heme iron complexes. Conceptually, these aziridination methods could provide insight into differential reactivity of oxygen- and nitrogen-transfer intermediates. Practically, aziridination protocols are synthetically important: aziridines are potent pharmacophores, ligands for catalysis, and versatile precursors to bioactive fine chemicals and materials. Still, the most synthetically promising non-heme iron catalysts with two available coordination sites require an excess of alkene to produce racemic aziridines. We have investigated the influence of non-heme iron complex architecture on a mild aziridination process that employs a limiting quantity of olefin substrate (Figure 3).
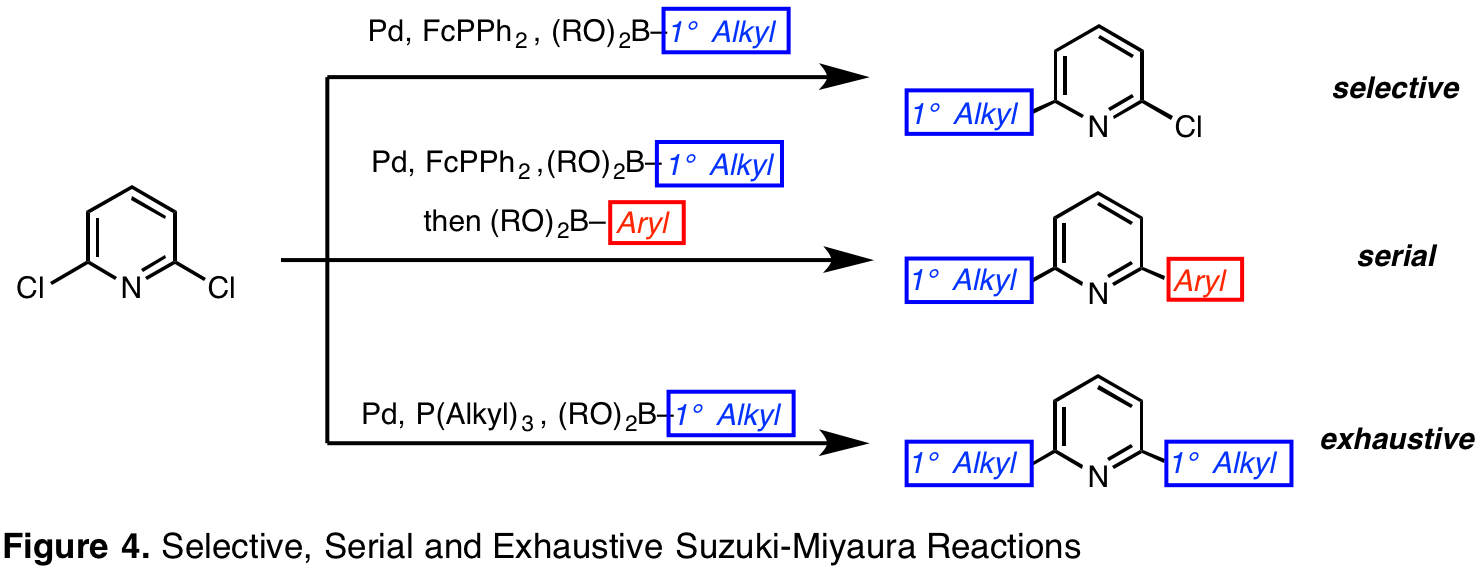
Selective, Serial and Exhaustive Protocols Enable Suzuki-Miyaura Reactions of 2,6-Dihalopyridines with Alkylpinacol Boronic Esters. Initially, this series of methods was investigated during the pursuit of access a catalyst scaffold for use in atom-transfer reactions. The anticipated route would generate a pyridine-centered ligand based on mild cross-coupling strategies. We focused on Suzuki-Miyaura conditions because this class of reactions offers practical advantages, including mild reaction conditions, commercially available reagents with low toxicity, and broad functional group tolerance. Nevertheless, few conditions could be used to cross-couple alkyl organoboron compounds with aryl halides, especially 2-pyridyl halides. To address this gap in the literature, we have developed and disclosed selective and serial Suzuki-Miyaura protocols (Org. Lett. 2016, 18, 4440-4443). Under the disclosed conditions, polyhalogenated six-membered heteroarenes engage in cross-coupling reactions with predictable selectivity, or react with distinct organoboron compounds to form two new carbon–carbon bonds in series in a single reaction vessel (Figure 4). As an extension of this research, a third set of conditions have been identified to facilitate the exhaustive reaction of 2,6-dihalopyridines with alkylpinacol boronic acids (Chem. Commun. 2017, 53, 7270-7273).
Impact of Funding on Co-workers. On the basis of publications describing the Suzuki-Miyaura cross-coupling technologies herein, the PI was a 2017 Thieme Chemistry Awardee. Moreover, this funding has enabled the PI to generate the preliminary results necessary to launch a research career.
Each of the talented individuals funded by this ACS PRF grant gained research experience with ligand synthesis, organometallic catalyst development, or methodology development. Importantly, this research grant provided stipend support Dr. S. Laulhe in his ultimate year as a postdoctoral scientist, and the research he carried out enabled him to secure a tenure-track faculty position in the Department of Chemistry and Chemical Biology at Indiana University-Purdue University Indiana. In addition, this grant partially supported the work of graduate students Mina Shehata (stipend) and J. Miles Blackburn (supplies), undergraduate students Ekaterina Khlystova (partial summer stipend) and Ashling Zhang (supplies). For Ms. Zhang, this support enabled her first synthetic research experiences as an independent study student.
In the course of this research, trainees acquired experience as scientific communicators. All trainees have presented their work in group meetings. At Duke, Ms. Zhang presented her ligand synthesis at the Spring 2016 Visible Thinking poster session, and Mr. Shehata presented a poster for the Chemistry Department Graduate Research Symposium and a talk at the fall 2017 ACS National Meeting. In more formal disclosures, the manuscripts to disclose Suzuki-Miyaura reactions of 2,6-dichloropyridines have been published, and presented his research at the Organic Reactions and Processes Gordon Research Conference. The guided chlorination reaction has been presented at three Gordon Research Conferences in the summer of 2017.










